 |
Introducción: Calibrar un espectrofotómetro
En el panorama de la ciencia analítica moderna, la espectrofotometría UV-Visible (UV-Vis) se erige como una técnica angular, indispensable en campos que van desde la química clínica y la vigilancia medioambiental hasta el control de calidad farmacéutico. Este potente método permite cuantificar con precisión las sustancias midiendo su absorción de la luz. Sin embargo, la precisión y fiabilidad de esta técnica -y la validez de los datos que produce- dependen por completo de un único proceso innegociable: la calibración.
La calibración es mucho más que una tarea rutinaria o un ejercicio de marcar casillas. Es el proceso fundamental que sustenta la integridad científica de cada medición, garantizando que un instrumento funcione según sus tolerancias especificadas. Para un laboratorio, la diferencia entre un espectrofotómetro correctamente calibrado y uno que se ha desviado de sus especificaciones es profunda. Puede significar la diferencia entre un resultado publicable y un artículo retirado, un lote de producto aprobado y una costosa retirada, o el cumplimiento de la normativa y un hallazgo crítico en una auditoría.
Esta guía constituye un recurso definitivo para directores de laboratorio, investigadores de alto nivel, profesionales de la garantía de calidad e importadores y distribuidores de instrumentos científicos. Profundiza en los principios básicos de la calibración de espectrofotómetros, explorando el “por qué” de su importancia, el “qué” de los parámetros críticos de rendimiento que deben comprobarse y el “cómo” de la aplicación de un programa de calibración sólido y conforme a la normativa. Proporcionaremos un marco paso a paso para crear un procedimiento operativo estándar (POE), orientación para seleccionar los materiales de referencia certificados (MRC) correctos y una guía práctica para solucionar los fallos más comunes. Al comprender y aplicar estos principios, los laboratorios pueden garantizar el máximo nivel de integridad de los datos, cumplir las estrictas exigencias normativas y maximizar el valor de su inversión en instrumentación analítica.
Sección 1: La base de una medición precisa: ¿Por qué calibrar su espectrofotómetro?
1.1 Más allá de las comprobaciones rutinarias: El papel de la calibración en la integridad y reproducibilidad de los datos
En esencia, la calibración es el proceso de verificar y ajustar el rendimiento de un instrumento a un patrón conocido y trazable. De este modo se establece un punto de referencia fundamental que garantiza que el espectrofotómetro mide lo que debe medir con un alto grado de precisión. Con el tiempo, todos los instrumentos analíticos están sujetos a desviaciones debidas a factores como el envejecimiento de los componentes (especialmente las fuentes de luz), los cambios en la temperatura y la humedad ambiente y el desgaste general. La calibración es el proceso formal que identifica y corrige estas desviaciones, garantizando que los datos generados no sólo son precisos en un momento dado, sino que también son reproducibles a largo plazo y comparables entre distintos instrumentos y laboratorios.
Además, una calibración adecuada garantiza la trazabilidad, un concepto crítico en metrología. Al utilizar materiales de referencia certificados (CRM) cuyos valores están vinculados a patrones primarios de institutos nacionales de metrología como el Instituto Nacional de Normas y Tecnología (NIST), la calibración crea una cadena ininterrumpida de comparaciones. Esto proporciona pruebas objetivas y científicamente defendibles de la precisión de un instrumento, una piedra angular de la gestión de la calidad y el cumplimiento de la normativa.
1.2 Cumplimiento de las exigencias normativas: Cumplimiento de farmacopeas y normas de calidad
En las industrias reguladas, especialmente en el sector farmacéutico, la calibración de espectrofotómetros no es simplemente una práctica recomendada, sino un requisito obligatorio. Los marcos de control de calidad como la norma ISO 9001, las Buenas Prácticas de Laboratorio (BPL) y las Buenas Prácticas de Fabricación (BPF) exigen la comprobación y validación periódicas del funcionamiento de los instrumentos analíticos para garantizar que se ajustan a su finalidad.
Las principales farmacopeas internacionales proporcionan orientación explícita sobre los parámetros y criterios de aceptación para la calibración de espectrofotómetros UV-Vis. Entre los capítulos clave se incluyen el capítulo general de la Farmacopea de Estados Unidos (USP) “Espectroscopia ultravioleta-visible” y el capítulo 2.2.25 de la Farmacopea Europea (Ph. Eur.) “Espectrofotometría de absorción, ultravioleta y visible”. Estas normas no son estáticas; se revisan y actualizan periódicamente. Por ejemplo, la USP sufrió importantes revisiones que entraron en vigor en diciembre de 2022, introduciendo requisitos más estrictos para la validación estadística, como la realización de múltiples mediciones replicadas para determinadas pruebas. El cumplimiento de las versiones actuales de estas farmacopeas es esencial para cualquier laboratorio que participe en el desarrollo, la fabricación o el control de calidad de productos farmacéuticos.
1.3 El impacto financiero y científico de la inexactitud
Considerar el calibrado únicamente como una tarea técnica pasa por alto su profunda importancia estratégica. Se trata de una estrategia proactiva de gestión de riesgos que protege contra los fallos en cascada originados por una sola medición inexacta. Un instrumento no calibrado o mal calibrado produce datos poco fiables, que introducen errores sistémicos en cada análisis y decisión posteriores.
Las consecuencias de esta cadena de fallos son graves. En un entorno de control de calidad, puede conducir a resultados fuera de especificación (OOS), desencadenando costosas investigaciones, lotes de productos fallidos, retrasos en la fabricación y, en el peor de los casos, retiradas de productos que conllevan inmensos daños financieros y de reputación. En un contexto de investigación y desarrollo, los datos inexactos pueden dar lugar a conclusiones científicas inválidas, al despilfarro de recursos en la búsqueda de pistas falsas y a la retractación de trabajos publicados. Para los importadores y distribuidores de instrumentos, la capacidad de demostrar que un instrumento puede calibrarse y validarse de forma fiable según las normas mundiales es un factor crítico para su comerciabilidad y una salvaguarda contra la responsabilidad. Por lo tanto, la inversión en un programa de calibración sólido -incluido el coste de los CRM de alta calidad y el tiempo de los técnicos- es una póliza de seguro esencial contra los costes mucho mayores de los fallos analíticos.
Sección 2: Comprender los parámetros básicos de calibración
Una calibración completa de un espectrofotómetro implica la evaluación de varios parámetros clave de rendimiento. Cada parámetro evalúa un aspecto diferente de los sistemas óptico y electrónico del instrumento, y juntos proporcionan una imagen completa de su rendimiento. La comprensión de estos parámetros es el primer paso hacia una calibración y localización de averías eficaces.
2.1 Precisión de la longitud de onda: ¿Está midiendo en la dirección correcta?
Definición: La precisión de la longitud de onda es la capacidad del instrumento para seleccionar e informar correctamente sobre una longitud de onda de luz específica. En términos sencillos, verifica que el eje x de la salida del espectrofotómetro es correcto.
Por qué es importante: Todo el principio de la espectrofotometría se basa en la medición de la absorbancia en longitudes de onda específicas en las que un analito absorbe la luz. Si el ajuste de la longitud de onda del instrumento es inexacto -por ejemplo, si está ajustado a 280 nm pero en realidad emite luz a 282 nm- todas las mediciones posteriores serán erróneas. Esto puede dar lugar a errores significativos en el análisis cuantitativo, sobre todo cuando se mide en la pendiente pronunciada de un pico de absorción, y puede provocar la identificación errónea de compuestos en el análisis cualitativo.
Causas de inexactitud: Los errores de longitud de onda suelen deberse a desplazamientos mecánicos u ópticos dentro del monocromador. Esto puede deberse a golpes físicos, vibraciones, expansión y contracción térmica de los componentes, o a la desalineación gradual de la rejilla de difracción o los espejos con el paso del tiempo. El envejecimiento de la fuente de luz del instrumento, como una lámpara de deuterio, también puede hacer que sus líneas de emisión características se desplacen, afectando a las comprobaciones de calibración internas.
2.2 Precisión fotométrica y linealidad: ¿Es la respuesta del instrumento verdadera y proporcional?
Definición: La exactitud fotométrica es la capacidad del detector y la electrónica del instrumento para medir correctamente la cantidad de luz que atraviesa una muestra e informar de un valor de absorbancia (o transmitancia) lo más cercano posible al valor verdadero. La linealidad fotométrica, un concepto estrechamente relacionado, evalúa si la respuesta fotométrica del instrumento es directamente proporcional a la concentración del analito en un intervalo especificado. Esta proporcionalidad es la base de la ley de Beer-Lambert, expresada como
A=ϵcb, donde A es la absorbancia, ϵ es la absortividad molar, c es la concentración y b es la longitud del camino.
Por qué son importantes: Estos parámetros son el corazón del análisis cuantitativo. Cualquier error en la precisión fotométrica se traduce directamente en un error en la concentración calculada de la muestra. Si la respuesta del instrumento no es lineal, la curva de calibración construida a partir de soluciones patrón no será válida, lo que imposibilitará determinar la concentración de una muestra desconocida de forma fiable.4 Cabe señalar que, mientras que la Farmacopea Europea especifica una prueba de linealidad distinta, la USP actualizada
ha evolucionado hacia un enfoque en el que la linealidad se verifica implícitamente probando la precisión fotométrica en múltiples niveles de absorbancia en todo el rango operativo del instrumento.
2.3 Luz parásita: La cuantificación del ruido no deseado
Definición: La luz parásita es cualquier luz no deseada o “falsa” que llega al detector pero que se encuentra fuera de la banda de longitud de onda específica seleccionada por el monocromador.
Por qué es importante: La luz parásita es una de las fuentes de error más importantes en espectrofotometría, sobre todo en el caso de las muestras de alta concentración que tienen valores de absorbancia elevados. Introduce un error sistemático que provoca una desviación negativa de la ley de Beer-Lambert, haciendo que la absorbancia medida parezca inferior a su valor real. Este efecto limita efectivamente el extremo superior del rango dinámico utilizable del instrumento, ya que la señal constante de la luz parásita se hace más significativa a medida que disminuye la luz transmitida verdadera.
Causas: La luz parásita puede tener su origen en varias fuentes, incluidas las imperfecciones y la dispersión de las superficies ópticas como las rejillas y los espejos, las fugas de luz del entorno circundante al instrumento y los reflejos internos dentro de la carcasa del monocromador (un fenómeno conocido como espectros reentrantes).
2.4 Resolución espectral: Distinción entre vecinos
Definición: La resolución espectral, o poder de resolución, es la capacidad de un espectrofotómetro para distinguir entre dos características espectrales adyacentes, como dos picos de absorción muy próximos. Esta capacidad viene determinada fundamentalmente por el ancho de banda espectral (SBW) del instrumento, que es la anchura de la banda de longitudes de onda que pasa a través de la rendija de salida del monocromador.
Por qué es importante: Un instrumento con poca resolución puede no separar dos picos distintos, haciendo que aparezcan como un único pico amplio. También puede “aplanar” la altura real de las bandas de absorción nítidas, dando lugar a mediciones inexactas de los picos máximos y, en consecuencia, a errores en el análisis cualitativo y cuantitativo. La alta resolución es especialmente crítica para analizar mezclas complejas con espectros superpuestos o compuestos que presentan estructuras espectrales finas.
La interrelación entre estos parámetros es un concepto crucial para la gestión eficaz de los instrumentos. No son parámetros aislados; un fallo en uno puede causar directamente o imitar un fallo en otro. Por ejemplo, un fallo en una prueba de linealidad fotométrica está causado muy a menudo por un alto nivel de luz parásita. La ley de Beer-Lambert se basa en el uso de luz monocromática. La luz parásita introduce radiación policromática, violando este supuesto fundamental. En concentraciones de muestra elevadas, en las que la luz transmitida real es mínima, la señal de fondo constante procedente de la luz parásita se convierte en una gran proporción de la luz total que incide en el detector. Esto hace que la respuesta del instrumento se estanque, destruyendo la linealidad. Por lo tanto, un técnico que observe un fallo de linealidad debe investigar la luz parásita como causa principal antes de sospechar de un problema más complejo del detector. Del mismo modo, una resolución deficiente puede afectar negativamente a la precisión de la longitud de onda al ensanchar los picos y dificultar la identificación precisa de la verdadera longitud de onda de máxima absorbancia. Reconocer estos vínculos causales es clave para una resolución de problemas eficiente y eficaz.
Sección 3: El conjunto de herramientas esenciales: Selección y uso de materiales de referencia certificados (MRC)
Todo el proceso de calibración se basa en comparar el rendimiento del instrumento con un punto de referencia conocido. Estos puntos de referencia son los materiales de referencia certificados (CRM), es decir, patrones físicos o soluciones químicas con propiedades conocidas y documentadas con precisión. La selección y el uso adecuado de los CRM son fundamentales para una calibración válida y defendible.
3.1 El patrón oro: Por qué los MRC trazables a NIST no son negociables
Para que una calibración tenga sentido, los patrones utilizados deben ser de un orden de precisión superior al del instrumento que se está comprobando. Esto se consigue mediante el uso de CRM obtenidos de una fuente acreditada y reconocida. El atributo más crítico de un CRM es su trazabilidad. La trazabilidad NIST significa que el valor certificado del patrón se ha determinado mediante una cadena ininterrumpida de comparaciones con patrones físicos primarios mantenidos por el Instituto Nacional de Normas y Tecnología (NIST) u otro Instituto Nacional de Metrología equivalente.
Esta trazabilidad proporciona una prueba objetiva, verificable y legalmente defendible de la precisión de la medición. Al comprar MRC, es imprescindible asegurarse de que se suministran con un Certificado de Calibración formal que detalle los valores certificados, la incertidumbre de medición asociada, la fecha de certificación y la declaración de trazabilidad.
3.2 Estándares líquidos frente a sólidos: Una elección estratégica
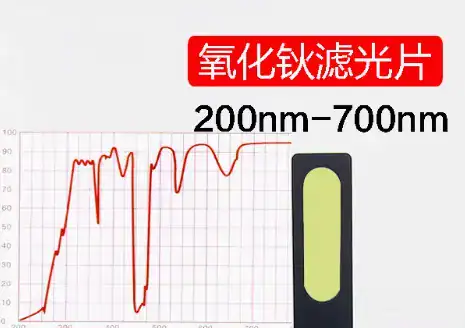
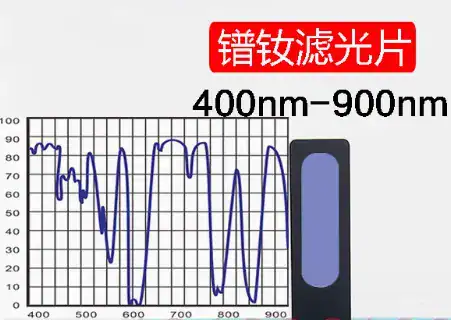
Los CRM para la calibración de espectrofotómetros están disponibles en dos formas principales: soluciones líquidas y filtros de estado sólido. La elección entre uno y otro es una decisión estratégica que repercute en el flujo de trabajo del laboratorio, la fiabilidad y el coste total de propiedad.
 |
 |
 |
- Estándares de estado sólido (por ejemplo, vidrio de óxido de holmio, vidrio de didimio, filtros de densidad neutra):
Ver Filtro de espectrofotómetro HINOTEK aquí.
- Ventajas: Estos patrones son apreciados por su excepcional estabilidad y durabilidad a largo plazo. Si se manipulan y almacenan correctamente, no se degradan y es posible que nunca requieran una recalibración. Son fáciles de usar, no requieren preparación, lo que elimina una fuente importante de errores del operario. Muchos están recubiertos para ser resistentes a los arañazos, lo que aumenta aún más su longevidad.
- Desventajas: Los filtros de estado sólido suelen tener un precio de compra inicial más elevado. También puede haber problemas de compatibilidad, ya que la forma física fija puede no ser adecuada para la geometría del haz de todos los modelos de espectrofotómetro. Además, la matriz de vidrio puede alterar ligeramente el espectro de absorción del material incrustado (como el óxido de holmio) en comparación con sus propiedades en disolución.
- Patrones líquidos (por ejemplo, dicromato de potasio, perclorato de holmio, cloruro de potasio):
- Ventajas: La principal ventaja de los patrones líquidos es que se miden de la misma manera que las muestras de rutina: como una solución en una cubeta patrón. Esto imita perfectamente las condiciones analíticas de un experimento típico. Muchos métodos de farmacopea exigen específicamente el uso de patrones líquidos.
- Desventajas: Los estándares líquidos tienen una vida útil limitada y pueden degradarse con el tiempo debido a reacciones químicas lentas e irreversibles. Son muy susceptibles a los errores del operador durante la preparación, incluidas las imprecisiones en el pesaje y la dilución, así como a la contaminación. Muchos de los productos químicos utilizados, como el dicromato potásico y el ácido perclórico, son peligrosos, por lo que requieren procedimientos cuidadosos de manipulación y eliminación.
Aunque los estándares líquidos pueden parecer más económicos a primera vista, un análisis más profundo revela importantes costes ocultos relacionados con el tiempo de preparación de los técnicos, el riesgo de error que obliga a repetir el trabajo, los costes de compra recurrentes debidos a la estabilidad limitada y la gestión de materiales peligrosos. Los patrones de estado sólido representan una mayor inversión inicial pero ofrecen un menor coste total de propiedad al simplificar el flujo de trabajo de calibración, reducir los gastos operativos a largo plazo y mejorar drásticamente la fiabilidad y reproducibilidad del proceso de calibración. Se trata de una poderosa propuesta de valor para cualquier director de laboratorio o agente de compras centrado tanto en el cumplimiento como en la eficiencia.
3.3 Tabla: CRM recomendados para cada parámetro de calibración
La siguiente tabla ofrece una lista consolidada de los CRM de uso común y recomendados por la farmacopea para cada uno de los parámetros básicos de calibración.
| Parámetro | CRM primario | Gama espectral | Descripción y uso | Farmacopeas clave |
| Precisión de la longitud de onda | Óxido de holmio (solución o filtro de vidrio) | UV/Vis (240-650 nm) | Proporciona múltiples picos nítidos y bien definidos para verificar el eje x. La solución es la preferida por la EP, pero el vidrio es más estable. | USP, EP |
| Óxido de didimio (filtro de vidrio) | Vis/NIR (400-880 nm) | Utilizado para la verificación de la longitud de onda en longitudes de onda más largas, exigido por la USP si se mide >650 nm. | USP | |
| Óxido de cerio (solución) | UV lejano (200-255 nm) | Recomendado para verificar el rendimiento en el rango UV bajo. | USP, EP | |
| Líneas de lámpara de deuterio (D2) o mercurio (Hg) | Líneas de emisión específicas (por ejemplo, D2: 486,0, 656,1 nm) | Utiliza las propias líneas de emisión de la lámpara del instrumento como patrón físico fundamental. A menudo se utiliza para comprobaciones internas/automatizadas. | USP, BP | |
| Precisión fotométrica | Solución de dicromato potásico (PDC) | UV (235-350 nm) | El patrón líquido más ampliamente reconocido para la exactitud fotométrica UV. Se utilizan concentraciones múltiples para comprobar la linealidad. | USP, EP, BP |
| Filtros de densidad neutra (ND) (vidrio o metal sobre cuarzo) | Vis/NIR (400-900+ nm) | Filtros estables y sólidos con absorbancia nominalmente plana en todo el espectro visible. El metal sobre cuarzo ofrece una gama más amplia. | USP | |
| Luz parásita | Soluciones/filtros de corte (por ejemplo, KCl, NaI, NaNO₂, acetona) | Rangos UV específicos | Estos materiales son opacos a la longitud de onda de ensayo. Cualquier luz detectada es luz parásita. La elección depende del rango de longitud de onda que se esté probando. | USP, EP, BP |
| Resolución espectral | Tolueno en solución de hexano | UV (~266-269 nm) | La relación entre el máximo de absorbancia (~269 nm) y el mínimo adyacente (~266 nm) se utiliza para evaluar la capacidad del instrumento para resolver características espectrales finas. | USP, EP, BP |
Sección 4: Guía paso a paso para la calibración de espectrofotómetros: Un marco SOP
Esta sección proporciona un marco detallado para un Procedimiento Operativo Estándar (SOP) para la calibración de espectrofotómetros UV-Vis. Los procedimientos se ajustan a los estrictos requisitos de las principales farmacopeas, en particular el capítulo .
4.1 Lista de comprobación previa a la calibración: Preparando el terreno para el éxito
Antes de realizar cualquier medición, deben completarse una serie de pasos preparatorios para garantizar la validez del proceso de calibración.
- Entorno: El espectrofotómetro (Encuentre el espectrofotómetro HINOTEK perfecto para su laboratorio) debe estar situado en un entorno operativo estable. Esto significa que debe estar sobre un banco robusto libre de vibraciones, alejado de la luz solar directa o de las corrientes de aire de los conductos de aire acondicionado, y en una habitación con temperatura y humedad controladas. El aire también debe estar limpio y libre de vapores químicos o humo que puedan interferir con la óptica.
- Estado del instrumento: Encienda el instrumento y deje que complete sus comprobaciones de autodiagnóstico. Fundamentalmente, deje que el instrumento se caliente durante el periodo especificado por el fabricante, que suele ser de al menos 30 a 60 minutos. Este periodo de calentamiento es esencial para la estabilización térmica de las fuentes de luz (por ejemplo, las lámparas de deuterio y tungsteno) y la electrónica del detector, garantizando una señal de salida estable.
- Limpieza: Un instrumento limpio es un requisito previo para obtener mediciones precisas. Inspeccione y limpie visualmente el exterior del instrumento y el compartimento de la muestra. Y lo que es más importante, asegúrese de que todas las cubetas y/o filtros de referencia sólidos estén impecablemente limpios. Manipúlelas sólo por sus lados esmerilados para evitar huellas dactilares en las superficies ópticas. Limpie las cubetas con un disolvente adecuado de grado espectroscópico y séquelas con un paño sin pelusas. Inspecciónelas cuidadosamente en busca de arañazos, astillas o residuos, ya que estos defectos dispersarán la luz e invalidarán las mediciones.
4.2 El primer paso universal: Realizar una corrección de la línea de base (blanking)
Toda medición espectrofotométrica es una medición comparativa. El instrumento mide la luz que pasa a través de la muestra y la compara con la luz que pasa a través de una referencia, o “blanco”. Este proceso, conocido como blanking o puesta a cero, es esencial para restar matemáticamente la señal de absorbancia de la propia cubeta y del disolvente en el que se disuelve la muestra. Esto garantiza que la absorbancia final notificada se debe únicamente al analito de interés.
Procedimiento: Llene una cubeta de cuarzo limpia y de alta calidad hasta las tres cuartas partes con la solución en blanco (por ejemplo, el ácido específico utilizado para disolver un patrón líquido o agua de gran pureza). Coloque la cubeta en el portamuestras del instrumento, asegurándose de que las ventanas ópticas transparentes estén alineadas con la trayectoria del haz de luz. Ejecute la función de corrección de la línea de base o “absorbancia cero” del instrumento. Cuando se utilizan filtros sólidos, la medición del blanco suele realizarse con un portamuestras vacío (es decir, un blanco de aire).
4.3 Procedimiento 4.1: Verificación de la precisión de la longitud de onda (según USP )
Este procedimiento verifica que la escala de longitud de onda del instrumento (eje x) es correcta.
- Selección del estándar: Seleccione un CRM con picos certificados que abarquen la gama de longitudes de onda operativa prevista del laboratorio. Para el trabajo general con UV-Vis, es habitual utilizar una solución de óxido de holmio o un filtro de vidrio. Si se realizan mediciones por encima de 650 nm, la USP también exige un filtro de didimio.
- Medición:
- Realice una corrección de la línea de base utilizando el blanco adecuado (por ejemplo, ácido perclórico 1,4 M para una solución de óxido de holmio, o aire para un filtro de vidrio).
- Coloque el patrón de longitud de onda certificado en el portamuestras.
- Configure el instrumento para que realice un barrido de longitudes de onda en el intervalo certificado del patrón.
- El software del instrumento identificará las longitudes de onda de máxima absorbancia para los distintos picos del espectro del patrón.
- Para los instrumentos de barrido (sin matriz de diodos), la USP exige al menos seis mediciones replicadas. Debe calcularse la media de estas seis mediciones para cada pico certificado.
- Evaluación:
- Precisión: Para cada pico, calcule la diferencia entre la longitud de onda media medida y el valor indicado en el certificado del CRM. Esta diferencia debe estar dentro de los criterios de aceptación especificados por la farmacopea pertinente (por ejemplo, ±1 nm en el rango UV para la USP).
- Precisión (repetibilidad): Para los instrumentos sin matriz de diodos, calcule la desviación estándar (DE) de las seis mediciones replicadas para cada pico. La SD no debe superar el límite de la farmacopea (por ejemplo, ≤ 0,5 nm para la USP).
4.4 Procedimiento 4.2: Verificación de la exactitud y repetibilidad fotométricas (según USP )
Este procedimiento verifica que la escala de absorbancia del instrumento (eje y) es correcta y reproducible.
- Selección de estándares: Seleccione al menos dos (y preferiblemente tres) CRM con diferentes valores de absorbancia que abarquen el rango operativo del laboratorio. Para el rango UV (<400 nm), utilice soluciones ácidas de dicromato potásico (PDC). Para el rango visible (>400 nm), utilice filtros sólidos de densidad neutra (ND).
- Medición:
- Ajuste el instrumento al modo de medición de longitud de onda fija en las longitudes de onda específicas certificadas para el patrón elegido (por ejemplo, 235 nm, 257 nm, 313 nm y 350 nm para PDC).
- Realice una medición en blanco utilizando el disolvente adecuado (por ejemplo, ácido perclórico 0,001 M para los estándares PDC).
- Mida la absorbancia del primer CRM. Según las directrices de la USP, realice al menos seis mediciones repetidas.
- Repita este proceso para cada uno de los otros CRM seleccionados.
- Evaluación:
- Precisión: Para cada estándar, calcule la media de las seis lecturas de absorbancia. Compare este valor medio con el valor certificado. La desviación debe estar dentro de los límites de aceptación.
- Repetibilidad (Precisión): Para cada estándar, calcule la desviación estándar de las seis mediciones replicadas. La DE debe estar por debajo del límite especificado.
4.5 Procedimiento 4.3: Medición de la luz difusa (según USP )
Este procedimiento cuantifica el nivel de radiación parásita en el instrumento a una longitud de onda específica.
- Selección del estándar: Elija una solución o filtro de corte de luz difusa que sea apropiado para la región de longitud de onda de interés. Se suele utilizar una solución al 1,2% p/v de cloruro potásico (KCl) en agua para realizar pruebas a 198 nm.
- Medición:
- Ajuste el instrumento a una longitud de onda fija de 198 nm.
- Utilice agua de gran pureza como referencia/blanco.
- Mida la absorbancia de la solución de KCl al 1,2%.
- Evaluación: La solución de KCl es esencialmente opaca a 198 nm. Por lo tanto, cualquier luz que se transmita y detecte se considera luz parásita. Una lectura de absorbancia alta corresponde a una baja transmisión y, por tanto, a una baja luz parásita. La absorbancia medida debe ser superior al límite especificado en la farmacopea (por ejemplo, > 2,0 unidades de absorbancia).
4.6 Procedimiento 4.4: Comprobación de la resolución espectral (según USP )
Este procedimiento evalúa la capacidad del instrumento para resolver detalles espectrales finos.
- Selección del estándar: Prepare una solución al 0,02% v/v de tolueno en hexano de grado espectroscópico.
- Medición:
- Utilice el disolvente hexano como referencia/blanco.
- Realice un barrido de longitudes de onda de la solución de tolueno desde aproximadamente 260 nm hasta 275 nm.
- A partir del espectro resultante, determine el valor de absorbancia en el pico máximo (que se produce aproximadamente a 269 nm) y el valor de absorbancia en la depresión adyacente, o mínimo (aproximadamente a 266-267 nm).
- Evaluación: Calcule la relación entre la absorbancia en el máximo y la absorbancia en el mínimo (Amax/Amin). Esta relación debe cumplir o superar el límite establecido por la farmacopea (por ejemplo, ≥ 1,3 para USP, ≥ 1,5 para BP/IP).
4.7 Tabla: USP y Ph. Eur. Resumen de criterios de aceptación
Esta tabla sirve como guía de referencia rápida para los criterios de aceptación clave definidos por las Farmacopeas de Estados Unidos y Europea.
| Parámetro | Farmacopea | Criterios de aceptación |
| Precisión de la longitud de onda | USP | UV (200-400 nm): ±1 nm; Vis (400-900 nm): ±2 nm |
| Ph. Eur. 2.2.25 | UV (<400 nm): ±1 nm; Vis (>400 nm): ±3 nm | |
| Repetibilidad de la longitud de onda (precisión) | USP (matriz sin diodos) | Desviación estándar ≤ 0,5 nm |
| Precisión fotométrica (PDC en ácido) | USP | ≤ 1,0 A: ±0,010 A; > 1,0 A: ±1,0% del valor certificado |
| Precisión fotométrica (filtros ND) | USP | ≤ 1,0 A: ±0,008 A; > 1,0 A: ±0,8% del valor certificado |
| Repetibilidad fotométrica (Precisión) | USP | Desviación estándar ≤ 0,005 A (para valores ≤ 1,0 A) o ≤ 0,5% (para valores > 1,0 A) |
| Luz parásita | USP / BP | Utilizando solución de KCl al 1,2% a ~198 nm, Absorbancia > 2,0 A |
| Resolución | USP | Relación tolueno en hexano (A269nm/A267nm) ≥ 1,3 |
| BP / IP | Relación tolueno en hexano (A269nm/A266nm) ≥ 1,5 |
4.8 La ventaja moderna: Aprovechar los flujos de trabajo de calibración automatizados
Los requisitos cada vez más estrictos de las farmacopeas, como el mandato de la USP de seis mediciones repetidas para las pruebas de exactitud y precisión, representan un cambio de paradigma significativo respecto a las antiguas comprobaciones de un solo punto. La atención se centra ahora en la solidez estadística, evaluando tanto la media (exactitud) como la desviación estándar (repetibilidad). Este cambio tiene una consecuencia crítica: hace que la calibración manual sea mucho más laboriosa, requiera más tiempo y sea susceptible al error humano.
Este reto pone de relieve la importante ventaja de los espectrofotómetros modernos que incorporan módulos de software automatizados de verificación del rendimiento (PV) o de cualificación de los instrumentos (IQ/OQ). Estos flujos de trabajo automatizados guían al operador a través de todo el proceso, realizan las mediciones replicadas necesarias, ejecutan todos los cálculos y generan un informe completo y trazable. Las ventajas son sustanciales:
- Reducción de errores: La automatización elimina el riesgo de errores de transcripción de datos y procedimientos manuales incoherentes.
- Ahorro de tiempo: Lo que podría llevar horas de trabajo manual puede completarse mucho más rápidamente.
- Cumplimiento mejorado: Los sistemas automatizados están diseñados para cumplir los últimos requisitos de la farmacopea, proporcionando una documentación segura y trazable adecuada para las auditorías reglamentarias, incluido el cumplimiento de la FDA 21 CFR Parte 11.
Para los laboratorios, esta capacidad automatizada no es sólo una característica de conveniencia; es una solución directa a un importante desafío de cumplimiento y eficiencia, reduciendo el riesgo y liberando el valioso tiempo de los técnicos.
Sección 5: Solución de los fallos de calibración más comunes: Una guía práctica
Incluso con un procedimiento cuidadoso, las pruebas de calibración pueden fallar. Una prueba fallida no siempre indica un fallo catastrófico del hardware. Más a menudo, la causa raíz es un problema simple y corregible. Un enfoque sistemático del diagnóstico puede ahorrar mucho tiempo, evitar llamadas innecesarias al servicio técnico y capacitar al personal del laboratorio para mantener sus instrumentos de forma eficaz.
5.1 Un enfoque sistemático para diagnosticar los errores de calibración
La metodología de resolución de problemas más eficaz sigue un embudo lógico de complejidad, empezando por las fuentes de error más sencillas y comunes antes de escalar a posibilidades más complejas y costosas. Esta secuencia de diagnóstico debería ser:
- Error de usuario y de procedimiento: Verifique que se ha seguido el procedimiento correcto. ¿Se utilizó el CRM correcto? ¿Se preparó y utilizó correctamente el blanco?
- Consumibles y manipulación de muestras: Examine las cubetas, los disolventes y los CRM. ¿Están las cubetas limpias y sin arañazos? ¿Son puros los disolventes? ¿Han caducado o se han degradado los CRM líquidos?
- Factores ambientales: Compruebe si se han producido cambios recientes en el entorno del laboratorio. ¿Hay fluctuaciones de temperatura o vibraciones?
- Rendimiento y mantenimiento de los instrumentos: Revise el estado básico del instrumento. ¿Está la lámpara cerca del final de su vida útil? ¿Hay signos visibles de polvo o contaminación en el compartimento de muestras?
- Mal funcionamiento del hardware: Si se han agotado todas las demás posibilidades, es probable que se trate de un componente básico del hardware, como una óptica desalineada o un detector defectuoso, lo que requiere un servicio técnico.
5.2 Tabla: Guía de resolución de problemas de fallos de calibración
Esta tabla proporciona una guía práctica para diagnosticar y resolver los fallos más comunes en las pruebas de calibración.
| Síntoma / Fallo de la prueba | Posibles causas | Pasos de diagnóstico y soluciones |
| Falla la precisión de la longitud de onda (Los picos medidos se desvían de los valores certificados) | 1. El instrumento no es térmicamente estable: Los componentes no han alcanzado el equilibrio térmico. 2. CRM Incorrecto/Expirado: Utilización de un estándar incorrecto o que se ha degradado. 3. Desalineación óptica: La rejilla o los espejos se han desplazado debido a un choque físico. 4. Envejecimiento de la fuente de luz: Las líneas de emisión características de la lámpara de deuterio se han desplazado, o la contaminación por hidrógeno está causando interferencias. 5. Anchura de la rendija / Ancho de banda espectral incorrecto: El ajuste de SBW es demasiado amplio, lo que ensancha el pico y puede desplazar el máximo aparente. |
1. Solución: Asegúrese de que el instrumento se ha calentado durante el tiempo recomendado por el fabricante (por ejemplo, 30-60 minutos). Compruebe y mitigue las fuentes de fluctuaciones de temperatura en el laboratorio. 2. Solución: Compruebe que se ha utilizado el CRM correcto y verifique su certificado de calibración. Si utiliza un estándar líquido, asegúrese de que no ha superado su fecha de caducidad. Pruebe con un estándar certificado nuevo o diferente. 3. Solución: Ejecute la rutina de diagnóstico interno o de autocalibración del instrumento, si dispone de ella. Si el fallo persiste, el instrumento requiere una realineación por parte de un ingeniero de servicio cualificado. 4. Solución: Compruebe las horas de uso de la lámpara en el software del instrumento. Si está cerca del final de su vida útil, programe su sustitución por un técnico de servicio. 5. Solución: Verifique que el ajuste de SBW utilizado para la prueba coincide con los requisitos del método y las condiciones en las que se certificó el CRM. |
| Fallos de exactitud/linealidad fotométrica (La absorbancia se desvía del valor certificado, especialmente en absorbancia alta) | 1. Luz parásita alta: Esta es la causa más frecuente de falta de linealidad fotométrica en valores altos de absorbancia. 2. Cubetas sucias/arañadas/desajustadas: Los arañazos dispersan la luz, mientras que los residuos pueden absorberla. El uso de cubetas diferentes para el blanco y la muestra introduce errores de longitud de trayecto y de desajuste óptico. 3. Blanco incorrecto: La solución del blanco está contaminada, se preparó a partir de un lote de disolvente diferente al del patrón o se ha evaporado. 4. Preparación incorrecta del patrón: Errores en el pesaje o la dilución al preparar estándares líquidos como el PDC. 5. Degradación o saturación del detector: El fotodetector deja de responder linealmente a la intensidad de la luz. |
1. Diagnóstico: Realice inmediatamente la prueba de luz parásita (procedimiento 4.3). Si esta prueba falla, solucionar el problema de la luz parásita es la prioridad. 2. Solución: Limpie meticulosamente todas las cubetas con disolvente de grado espectroscópico y una toallita sin pelusa. Inspeccione en busca de arañazos bajo una luz brillante y deseche las que estén dañadas. Para obtener la máxima precisión, utilice siempre la misma cubeta de cuarzo de alta calidad para las mediciones del blanco y de la muestra. 3. Solución: Prepare un blanco nuevo utilizando exactamente el mismo disolvente utilizado para el patrón. Vuelva a poner en blanco el instrumento inmediatamente antes de medir el patrón. 4. Solución: Prepare un nuevo juego de patrones, prestando rigurosa atención a las técnicas analíticas de pesaje y volumetría. Para eliminar por completo esta variable, utilice un filtro de densidad neutra de estado sólido para la verificación. 5. Solución: Si se han descartado sistemáticamente todas las demás causas potenciales, esto apunta a un problema de hardware que requiere el diagnóstico de un ingeniero de servicio. |
| La prueba de luz parásita falla (la absorbancia es inferior al límite requerido, por ejemplo, < 2,0 A) | 1. Fugas de luz: La tapa del compartimento de muestras no está cerrada o no sella correctamente, o hay daños en la carcasa del instrumento que permiten la entrada de luz ambiental. 2. Óptica interna degradada/sucia: Una película de polvo o vapor químico se ha depositado en los espejos internos, las lentes o la rejilla, provocando una dispersión excesiva de la luz. 3. Rejilla de difracción imperfecta: La propia rejilla tiene defectos de fabricación que provocan la dispersión. Se trata de una propiedad inherente al instrumento. 4. Interferencia de la luz ambiente: La iluminación superior extremadamente brillante puede filtrarse a veces en el instrumento. |
1. Diagnóstico: Asegúrese de que la tapa del compartimento de muestras esté completamente cerrada y encaje correctamente. Compruebe que no haya daños visibles en la carcasa del instrumento. Pruebe a realizar la prueba en una habitación completamente a oscuras para ver si mejora el resultado. 2. Solución: Esto requiere un servicio profesional. No intente abrir la carcasa del instrumento para limpiar la óptica interna, ya que puede provocar una desalineación y daños adicionales. 3. Solución: Se trata de una limitación del diseño del instrumento, no de un fallo que pueda solucionarse. Los instrumentos con monocromadores dobles y rejillas holográficas de alta calidad tienen niveles de luz parásita inherentemente más bajos. Se trata de una especificación crítica a tener en cuenta a la hora de adquirir un instrumento. 4. Solución: Atenúe las luces de la sala durante la prueba para descartar esta sencilla fuente de interferencias. |
| La prueba de resolución falla (la relación tolueno/hexano está por debajo del límite requerido) | 1. Ancho de banda espectral (SBW) incorrecto: El SBW está ajustado demasiado ancho, lo que impide que el instrumento resuelva el pico y la depresión estrechamente espaciados. 2. Desalineación óptica: Los componentes del monocromador (rejilla, rendijas) no están correctamente alineados, lo que degrada el poder de resolución del instrumento. 3. Problema de hardware: Un problema en el mecanismo de accionamiento de la rejilla o en el detector impide el funcionamiento correcto. |
1. Solución: Compruebe los parámetros del método del instrumento. Asegúrese de que se selecciona la SBW más estrecha disponible para la prueba, tal y como requiere el procedimiento. 2. Diagnóstico/Solución: Se trata de un problema importante de hardware. Ejecute las pruebas de diagnóstico internas disponibles. El instrumento requerirá realineación por un ingeniero de servicio. 3. Diagnóstico/Solución: Si se confirma que la alineación es correcta, esto indica un problema de hardware más grave que requiere un servicio profesional. |
Sección 6: Mantenimiento del máximo rendimiento: Frecuencia de calibración y documentación
Un programa de calibración satisfactorio no es un acontecimiento puntual, sino un proceso continuo de verificación y documentación. El establecimiento de una frecuencia de calibración adecuada y el mantenimiento de registros meticulosos son esenciales para garantizar el cumplimiento continuo y la fiabilidad de los instrumentos.
6.1 Establecimiento de un calendario de calibración basado en los riesgos: ¿Con qué frecuencia es suficiente?
La frecuencia requerida para la calibración es un punto común de confusión, con recomendaciones que van desde la diaria a la anual. El enfoque más eficaz es un programa escalonado, basado en el riesgo, que combine comprobaciones frecuentes del rendimiento con cualificaciones completas menos frecuentes.
- Nivel 1: Comprobaciones de funcionamiento diarias o por uso (normalización): Antes de iniciar un nuevo lote de análisis o a intervalos regulares (por ejemplo, cada ocho horas), debe realizarse una simple comprobación del rendimiento. Esto suele implicar la ejecución de una nueva corrección de la línea de base (blanco) y la medición de un único patrón de comprobación estable (como un filtro de densidad neutra) para confirmar que el instrumento no se ha desviado significativamente. Este proceso suele denominarse “normalización”.
- Nivel 2: Calibración periódica completa (cualificación): La cualificación completa y multiparamétrica descrita en la sección debe realizarse de forma periódica. La frecuencia exacta depende de una evaluación de riesgos del uso del instrumento:
- Entornos de alto riesgo / alto uso: En un laboratorio de CC farmacéutico regulado por las GMP en el que los datos repercuten directamente en la liberación del producto, puede ser necesaria una cualificación completa trimestral o semestral.
- Entornos de riesgo moderado / uso reducido: Para muchos laboratorios de investigación académica o de I+D industrial, una cualificación completa realizada anualmente suele ser suficiente, siempre que las comprobaciones de rendimiento se superen de forma consistente.
- Calibración activada por eventos: Siempre debe realizarse una cualificación completa después de cualquier acontecimiento significativo, como una reparación o mantenimiento importante (por ejemplo, sustitución de la lámpara), traslado del instrumento, o si las comprobaciones rutinarias de funcionamiento fallan repetidamente o muestran una tendencia anómala.
6.2 La importancia de un registro de calibración detallado para las auditorías y el control de calidad
El registro de calibración es algo más que un simple registro; es la prueba principal que demuestra que un instrumento funcionaba dentro de sus especificaciones validadas en el momento en que se analizó una muestra concreta. Durante una auditoría reglamentaria realizada por organismos como la FDA o una evaluación ISO, el registro de calibración será uno de los primeros documentos solicitados.
Más allá de la conformidad, una documentación meticulosa crea un historial de rendimiento del instrumento. Estos datos históricos tienen un valor incalculable para el análisis de tendencias a largo plazo y para la resolución de problemas. Al transformar el registro de calibración de un registro estático en una herramienta dinámica y predictiva, el responsable de un laboratorio puede elevar su programa de calidad. Trazar los resultados clave de la calibración a lo largo del tiempo -como la desviación de un pico de longitud de onda específico o la absorbancia de un patrón de comprobación- puede revelar una deriva gradual y constante. Esta tendencia puede predecir un fallo inminente de un componente.
antes de que dé lugar a un evento crítico fuera de tolerancia, lo que permite programar un mantenimiento proactivo, maximizando el tiempo de funcionamiento del instrumento y evitando costosos fallos analíticos.
6.3 Plantilla: Campos esenciales para el registro de calibración de un espectrofotómetro
Un registro de calibración eficaz debe ser exhaustivo e inequívoco. La siguiente plantilla esboza los campos esenciales para un registro conforme y útil, que puede adaptarse al PNT oficial de un laboratorio.
REGISTRO DE CALIBRACIÓN DEL ESPECTROFOTÓMETRO:
Detalles del instrumento:
- ID del instrumento / Número de activo: _______________
- Modelo del instrumento: _______________
- Número de serie: _______________
- Ubicación (laboratorio/sala): _______________
Registro de eventos de calibración:
- Fecha de calibración: AAAA-MM-DD
- Próxima calibración prevista: AAAA-MM-DD
- Calibrado por (Nombre/Iniciales): _______________
- Tipo de calibración: [ ] Calibración anual [ ] Trimestral [ ] Post-mantenimiento [ ] Otro: _______________
Resultados de la prueba de parámetros:
| Parámetro Probado | CRM utilizado (nombre, nº de lote, nº de certificado) | Valor(es) certificado(s) | Valor(es) medido(s) (Media) | Criterios de aceptación | Desviación | DE (si procede) | Estado (Pasa/Falla) |
| Precisión de la longitud de onda | |||||||
| Precisión fotométrica | |||||||
| Luz parásita | |||||||
| Resolución | |||||||
| (Añada filas según sea necesario para cada estándar/longitud de onda probada) |
Resumen general:
- Estado global de la calibración: [ ] APROBADO [ ] SUSPENSO
- Comentarios / Medidas adoptadas: (por ejemplo, “El instrumento superó todas las pruebas” o “La precisión de la longitud de onda falló inicialmente. Ejecutó diagnósticos internos y volvió a realizar la prueba. Aprobado en el segundo intento. Véase el informe de diagnóstico adjunto”).
- Revisado por (Nombre/Título): _______________
- Fecha de revisión: AAAA-MM-DD
Conclusión: La calibración como piedra angular de la calidad y la confianza
La calibración rigurosa, periódica y bien documentada de un espectrofotómetro UV-Vis no es un impedimento para el flujo de trabajo del laboratorio, sino su mayor potenciador. Es la práctica disciplinada que transforma una sofisticada pieza de equipo óptico de un simple dispositivo de medición de luz en un instrumento científico validado, fiable y conforme.
Una calibración adecuada proporciona una confianza inquebrantable en cada punto de datos generado. Garantiza que los resultados sean precisos, reproducibles y defendibles, cumpliendo las normas mundiales más estrictas establecidas por las farmacopeas y los organismos reguladores. En última instancia, es el proceso que salvaguarda la integridad científica y financiera de todo el laboratorio. Al adoptar la calibración no como una carga sino como una piedra angular de la calidad, los investigadores y los fabricantes pueden garantizar que su trabajo resiste los niveles más altos de escrutinio, impulsando la innovación y ofreciendo productos seguros y eficaces al mercado.
Para las organizaciones que buscan instrumentos diseñados con la conformidad y la fiabilidad en su núcleo, y para la asociación de expertos en el establecimiento de protocolos de calibración robustos, HINOTEK proporciona tanto la tecnología como la experiencia para lograr una integridad de datos sin precedentes.
Si desea saber más sobre el espectrofotómetro, consulte nuestra página: ¿Qué es un espectrofotómetro y cómo funciona? La guía definitiva
El mantenimiento de esta guía corre a cargo del equipo técnico principal de HINOTEK, compuesto por ingenieros superiores y científicos de aplicaciones con más de dos décadas de experiencia práctica en campos como la microscopía, la centrifugación y la espectrofotometría. Nos comprometemos a garantizar que cada dato de esta guía -desde los principios de los instrumentos y las especificaciones técnicas hasta los consejos para la adquisición de equipos de laboratorio- mantenga el máximo nivel de precisión y actualidad.
Este contenido se revisa y actualiza periódicamente para reflejar los últimos estándares de la industria y los avances tecnológicos. Valoramos los comentarios de la comunidad científica mundial. Si tiene alguna pregunta o sugerencia, o desea comentar algún detalle técnico, no dude en ponerse en contacto con nuestro equipo de expertos en [email protected].