El mundo invisible en un haz de luz: Una guía completa para la medición de ácidos nucleicos y proteínas mediante espectrofotómetro
|
|
|


Introducción: El caballo de batalla del laboratorio de biología molecular
En el panorama de la biología molecular moderna, pocos instrumentos son tan ubicuos e indispensables como el espectrofotómetro. Desde un investigador que purifica una enzima novedosa hasta un técnico que realiza un control de calidad rutinario de las extracciones de ADN, este instrumento es un caballo de batalla fundamental que proporciona datos críticos sobre la concentración y la pureza de las moléculas invisibles de la vida. Es una tecnología angular que sustenta una amplia gama de aplicaciones posteriores, como la PCR, la secuenciación del ADN, la clonación, el Western blot y la cinética enzimática. La precisión de estos experimentos posteriores, a menudo complejos y costosos, depende de la calidad de los datos iniciales obtenidos a partir de este dispositivo aparentemente sencillo.
Este artículo pretende servir de guía exhaustiva y accesible para estudiantes, técnicos de laboratorio e investigadores que se adentren en el mundo de la espectrofotometría. Desmitificará el funcionamiento interno del instrumento, profundizará en las aplicaciones específicas para la cuantificación de ácidos nucleicos y proteínas, explorará su uso en la monitorización del crecimiento microbiano y, lo que es más importante, proporcionará un marco práctico de buenas prácticas y resolución de problemas para garantizar la generación de datos precisos, fiables y reproducibles. Al comprender no sólo cómo utilizar un espectrofotómetro, sino por qué funciona de la manera en que lo hace, los usuarios pueden liberar todo su potencial y evitar los escollos comunes que pueden comprometer su investigación.
Parte 1: El espectrofotómetro: una mirada bajo el capó
Antes de aplicar el espectrofotómetro a muestras biológicas, es esencial comprender los principios básicos que rigen su funcionamiento. Este conocimiento fundacional transforma el instrumento de una “caja negra” en una herramienta analítica transparente y predecible.
1.1 El principio rector: cómo medimos lo invisible
En esencia, la espectrofotometría es la medición cuantitativa del modo en que una sustancia química absorbe y transmite la luz en una gama específica de longitudes de onda. El principio básico es notablemente intuitivo: las diferentes moléculas de una solución absorben la luz de longitudes de onda específicas en distintos grados. Haciendo brillar un haz de luz controlado a través de una muestra y midiendo la cantidad de luz que se absorbe, podemos deducir la concentración de la sustancia de interés.
 . |
Una analogía sencilla puede encontrarse en un vaso de té o bebida coloreada. Si hace pasar una linterna a través de un vaso de té muy débil, la mayor parte de la luz pasará al otro lado. Sin embargo, si hace brillar la misma luz a través de un vaso de té muy fuerte y oscuro, el líquido absorberá mucha más luz y saldrá mucha menos. La “oscuridad” del té es análoga a la concentración de moléculas, y la cantidad de luz que se “come” la solución es análoga a su absorbancia. La espectrofotometría simplemente formaliza y cuantifica este fenómeno cotidiano con gran precisión.
1.2 Anatomía de un espectrofotómetro: El camino de la luz
Un espectrofotómetro se compone de varios componentes clave que trabajan en concierto para guiar la luz desde su fuente, a través de la muestra y hasta un detector. Comprender este recorrido de la luz es crucial para apreciar cómo se realizan las mediciones y qué puede salir mal.
 |
|
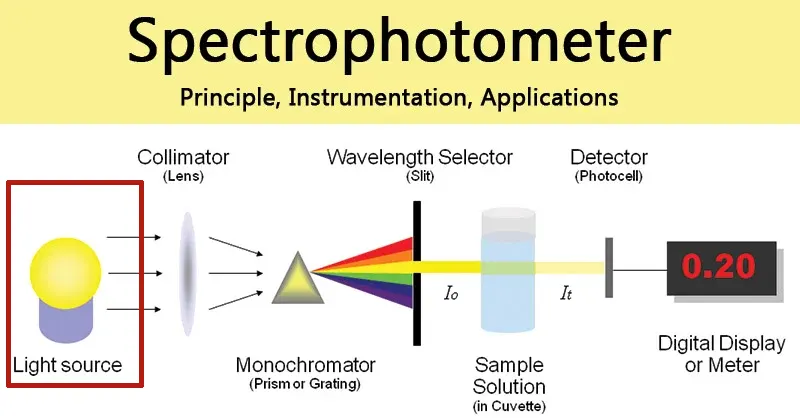
- Fuente de luz: El recorrido comienza con una lámpara que produce un amplio espectro de luz. Normalmente, los instrumentos utilizan una combinación de lámparas para cubrir toda la gama ultravioleta (UV) y visible. Se utiliza una lámpara de deuterio para la región UV (aproximadamente 190-400 nm), crítica para el análisis de ácidos nucleicos y proteínas, mientras que una lámpara halógena de tungsteno cubre las regiones visible e infrarroja cercana (aproximadamente 400-1100 nm).
- Monocromador: Este componente es el “selector de longitud de onda” y es posiblemente la parte más sofisticada del instrumento. La luz de amplio espectro de la lámpara entra en el monocromador e incide en un elemento dispersivo, que puede ser un prisma o, más comúnmente, una rejilla de difracción. Este elemento funciona como un prisma que divide la luz blanca en un arco iris, separando la luz en sus longitudes de onda constituyentes. Al girar con precisión la rejilla de difracción, sólo se permite que una banda muy estrecha de una longitud de onda única y específica (por ejemplo, 260 nm) atraviese una rendija de salida y se dirija hacia la muestra.
- Compartimento de la muestra y cubeta: El haz de luz seleccionado pasa al compartimento de la muestra, donde ésta se mantiene en un recipiente transparente llamado cubeta. Una cubeta estándar es un recipiente pequeño y rectangular diseñado para tener una longitud de trayectoria de luz precisa, que es la distancia que recorre la luz a través de la muestra. Esta longitud de trayectoria suele normalizarse en 1 cm para garantizar la coherencia y la comparabilidad de las mediciones entre instrumentos.
- Detector: Tras atravesar la muestra, la luz que no haya sido absorbida incide en un detector, como un tubo fotomultiplicador (PMT) o un fotodiodo. Este componente altamente sensible convierte los fotones de luz entrantes en una señal eléctrica. La intensidad de esta señal es directamente proporcional a la intensidad de la luz que incide sobre él.
- Visualización: La señal eléctrica del detector se envía a un microprocesador, que la convierte en un valor digital que se muestra al usuario, normalmente como transmitancia o absorbancia.
Una distinción clave en el diseño de los instrumentos es entre las configuraciones de haz único y las de haz doble. Un espectrofotómetro de haz simple mide secuencialmente la intensidad luminosa de una solución de referencia (el “blanco”) y de la solución de muestra. Un espectrofotómetro de doble haz, como el espectrofotómetro de doble haz HINOTEK L7, divide el haz de luz del monocromador en dos trayectorias separadas. Una trayectoria atraviesa la cubeta de referencia y la otra atraviesa simultáneamente la cubeta de muestra.6 Este diseño ofrece una ventaja significativa: proporciona una corrección en tiempo real de cualquier fluctuación en la salida de la lámpara o de los cambios en la atmósfera, lo que da como resultado una línea de base mucho más estable y una mayor precisión, en particular para las mediciones realizadas durante un período prolongado, como las exploraciones de cinética enzimática.
Tabla 1: Componentes clave de un espectrofotómetro y sus funciones
| Componente | Función | Analogía en el mundo real |
| Fuente de luz | Genera un amplio espectro de luz (UV y visible). | Una potente linterna multicolor. |
| Monocromador | Selecciona una única longitud de onda específica de luz para que atraviese la muestra. | Un prisma que aísla un color específico (por ejemplo, el rojo puro) de un arco iris. |
| Cubeta | Recipiente estandarizado y transparente que contiene la muestra con una longitud de recorrido fija. | Un vaso de té con una anchura específica y conocida. |
| Detector | Mide la intensidad de la luz que atraviesa la muestra y la convierte en una señal eléctrica. | Un medidor de luz que mide el brillo de la luz que no ha sido bloqueada por el té. |
| Pantalla | Convierte la señal eléctrica en un valor legible por el usuario (absorbancia). | La pantalla digital del fotómetro que muestra la lectura final. |
1.3 El lenguaje de la luz: Absorbancia y Transmitancia
Los espectrofotómetros informan de las mediciones de dos formas principales: transmitancia y absorbancia. Aunque están relacionadas, los científicos utilizan casi exclusivamente la absorbancia para el análisis cuantitativo.
- La transmitancia (T) es la fracción de la luz original que atraviesa con éxito la muestra. Se calcula como la relación entre la intensidad de luz transmitida (I) y la intensidad de luz incidente (I0). A menudo se expresa en porcentaje.T=I0/I
- La absorbancia (A), a veces denominada densidad óptica (DO), es la cantidad de luz que absorbe la muestra. Está relacionada con la transmitancia mediante una relación logarítmica negativa.10A=-log10(T)=-log10(I0I)
La razón por la que la absorbancia es la unidad preferida es fundamental para el análisis cuantitativo. La relación entre la transmitancia de una muestra y su concentración es logarítmica (y, por tanto, no lineal). En cambio, la relación entre la absorbancia y la concentración es directamente lineal. Esta relación lineal, descrita por la ley de Beer-Lambert, hace que sea mucho más sencillo crear curvas de calibración y calcular con precisión la concentración de una muestra desconocida. Una absorbancia de 0 corresponde a una transmitancia del 100% (no se absorbe luz), mientras que una absorbancia de 1 corresponde a una transmitancia del 10% (se absorbe el 90% de la luz).
1.4 La ley de Beer-Lambert: El corazón matemático de la espectrofotometría
La ley de Beer-Lambert (o ley de Beer) es la sencilla pero poderosa ecuación que constituye la base matemática de la espectrofotometría. Establece que la absorbancia de una solución es directamente proporcional a su concentración y a la longitud del recorrido de la luz a través de ella. La ley se expresa como
A=ϵlc Donde:
- A es la absorbancia medida por el instrumento (es un valor sin unidades).
- ϵ (épsilon) es la absortividad molar o coeficiente de extinción. Se trata de una constante que es una propiedad intrínseca de la molécula que se mide. Describe la intensidad con la que esa sustancia absorbe la luz a una longitud de onda específica. Sus unidades (por ejemplo, L⋅mol-1⋅cm-1) son tales que anulan las unidades de longitud de trayectoria y concentración, dejando la absorbancia sin unidades.
- l es la longitud de recorrido de la luz a través de la muestra, que viene determinada por la anchura de la cubeta. Casi siempre se estandariza a 1 cm.
- c es la concentración de la sustancia en la solución (por ejemplo, en mol⋅L-1). Este es el valor que los investigadores suelen querer determinar.
Aunque elegante, la ley de Beer-Lambert no es infalible. Su relación lineal es válida para la mayoría de los trabajos rutinarios de laboratorio, pero empieza a fallar a concentraciones muy elevadas. Cuando las moléculas están demasiado juntas, sus interacciones electrostáticas pueden alterar su capacidad para absorber la luz. Además, todos los espectrofotómetros tienen una pequeña cantidad de “luz parásita”, es decir, luz no deseada de otras longitudes de onda que llega al detector. En absorbancias altas, en las que se transmite muy poca luz de la longitud de onda correcta, esta luz parásita se convierte en una fracción significativa de la luz total que llega al detector, provocando que la absorbancia medida sea artificialmente baja y se desvíe de la linealidad. Por esta razón, las lecturas de absorbancia por encima de un determinado umbral (generalmente entre 1,5 y 2,0) se consideran poco fiables, y dichas muestras deben diluirse en el rango lineal para obtener una medición precisa.
Parte 2: Cuantificación de ácidos nucleicos – La piedra angular de la biología molecular
El uso más frecuente de un espectrofotómetro en un laboratorio de biología molecular es para la cuantificación de ácidos nucleicos. Ya se trate de preparar ADN para una reacción de PCR, ARN para un Northern blot u oligonucleótidos para la síntesis de genes, conocer la concentración y pureza precisas de la muestra es el primer paso y el más crítico.
2.1 La longitud de onda de 260 nm: La firma única del ADN y el ARN
La capacidad de cuantificar los ácidos nucleicos con espectrofotometría UV se deriva de la estructura química de sus componentes básicos. Las bases nitrogenadas -adenina (A), guanina (G), citosina (C), timina (T) y uracilo (U)- son todas estructuras anulares aromáticas. Estos anillos contienen un sistema de electrones pi deslocalizados que absorben fácilmente la luz ultravioleta. Esta absorción es máxima a una longitud de onda de aproximadamente 260 nm. Este pico de absorción distinto sirve como “huella dactilar” espectroscópica única, lo que convierte a los 260 nm en la longitud de onda estándar universal para cuantificar el ADN y el ARN.
2.2 De la absorbancia a la concentración: Los números mágicos
Aprovechando la ley de Beer-Lambert (A=ϵlc), podemos determinar fácilmente la concentración de una muestra de ácido nucleico. Dado que la longitud del trayecto (l) está normalizada a 1 cm y que la absortividad molar (ϵ) se ha determinado empíricamente para diferentes tipos de ácidos nucleicos, la ecuación puede simplificarse. Para una longitud de paso estándar de 1 cm, una absorbancia medida de 1,0 a 260 nm (A260) corresponde a una concentración bien establecida de ácido nucleico. Estos factores de conversión establecidos son los “números mágicos” utilizados en los laboratorios de todo el mundo.
Tabla 2: Factores de conversión estándar para la cuantificación de ácido nucleico (a A260)
| Tipo de ácido nucleico | Concentración para A260=1,0 (en µg/mL) | Abreviatura común |
| ADN bicatenario | 50 µg/mL | dsADN |
| ADN monocatenario | 33 µg/mL | ssADN |
| ARN monocatenario | 40 µg/mL | ARNsc |
| Oligonucleótidos monocatenarios cortos | ~20-33 µg/mL | Oligos |
Los datos procedentes dela concentración para oligonucleótidos varían en función de la composición de las bases.
Para calcular la concentración de una muestra desconocida, basta con medir su absorbancia a 260 nm y aplicar la siguiente fórmula:
Concentración (µg/mL)=A260×Factor de conversión×Factor de dilución
Por ejemplo, si una muestra de dsADN se diluye 10 veces y da una lectura A260 de 0,25, su concentración original se calcularía como: 0,25×50 µg/mL×10=125 µg/mL.
2.3 Evaluación de la pureza: Las relaciones críticas A260/A280 y A260/A230
Un valor de concentración carece de sentido sin una evaluación de la pureza de la muestra. Un mantra en biología molecular es “basura entra, basura sale”. Si una muestra de ADN está contaminada, la lectura A260 se inflará artificialmente, lo que conducirá a una gran sobreestimación de la concentración real de ADN. Esto puede hacer que los experimentos posteriores, como la PCR o la secuenciación del ADN, fracasen debido a la adición de cantidades incorrectas de ADN molde. La espectrofotometría proporciona dos ratios críticos para diagnosticar los tipos más comunes de contaminación.
La relación A260/A280: La comprobación de proteínas
Las proteínas son un contaminante común en las preparaciones de ácidos nucleicos. Los aminoácidos aromáticos triptófano y tirosina, que se encuentran en la mayoría de las proteínas, tienen un fuerte pico de absorbancia UV a 280 nm. Al medir la absorbancia tanto a 260 nm como a 280 nm, la relación de estos dos valores (A260/A280) proporciona una evaluación rápida de la contaminación por proteínas.
- Para el dsADN puro, la relación A260/A280 esperada es de ~1,8 a 1,9.
- Para el ARN puro, la relación A260/A280 esperada es de ~2,0 a 2,1.
Una relación significativamente inferior a estos valores indica la presencia de proteína contaminante o fenol residual del proceso de extracción, ambos de los cuales absorben fuertemente a 280 nm.
La relación A260/A230: La comprobación orgánica y salina
La relación A260/A230 sirve como medida secundaria, pero igualmente importante, de la pureza. Muchos reactivos comunes de laboratorio utilizados en los kits de purificación de ácidos nucleicos absorben la luz a 230 nm o cerca de esta cifra. Entre ellos se encuentran las sales caotrópicas como el tiocianato de guanidina y el clorhidrato de guanidina, el fenol, el TRIzol, el EDTA y los carbohidratos arrastrados del material de origen.
- Para una muestra de ácido nucleico puro (tanto ADN como ARN), la relación A260/A230 esperada debe ser superior a 2,0, situándose a menudo en el intervalo de 2,0-2,4.
Una relación A260/A230 baja es una fuerte señal de alarma, que indica que la muestra está probablemente contaminada con sales residuales o disolventes orgánicos procedentes del proceso de purificación. Tales contaminantes pueden inhibir las reacciones enzimáticas en las aplicaciones posteriores.
Corregir el “ruido”: El papel del A320 en la medición de la turbidez
Un último control de calidad consiste en medir la absorbancia a 320 nm (A320). Ni los ácidos nucleicos ni los contaminantes químicos comunes absorben la luz en esta región. Por lo tanto, cualquier lectura de absorbancia a 320 nm se debe normalmente a la dispersión de la luz causada por partículas en la solución, como restos celulares, partículas insolubles o perlas magnéticas residuales de un kit de purificación. Es una medida de la turbidez, o “suciedad”, de la muestra. Una muestra limpia y pura debería tener un valor A320 muy próximo a cero.
Tabla 3: Interpretación de los índices de pureza del ácido nucleico
| Ratio | Valor ideal (ADN) | Valor ideal (ARN) | Una relación baja indica… | Una relación alta indica… |
| A260/A280 | ~1.8 – 1.9 | ~2.0 – 2.1 | Contaminación por proteínas o fenol. | Generalmente no es un problema; puede indicar contaminación por ARN en una preparación de ADN. |
| A260/A230 | > 2.0 | > 2.0 | Contaminación con sales (guanidina), disolventes orgánicos (fenol, etanol) o carbohidratos. | Puede indicar un error en el blanco o el uso de una solución de blanco inadecuada. |
2.4 Cuando las proporciones mienten: una mirada más profunda a los contaminantes y factores de confusión
Aunque los índices de pureza son poderosas herramientas de diagnóstico, no son infalibles y deben interpretarse con precaución y en su contexto. Varios factores pueden inducir a error al usuario, y un conocimiento más profundo de los mismos es la marca de un usuario experto.
En primer lugar, la fiabilidad de los índices de pureza depende en gran medida de la concentración de la muestra. Para las muestras diluidas (generalmente por debajo de 20 ng/µL), los valores de absorbancia a 230 nm y 280 nm son extremadamente bajos, a menudo acercándose al límite de detección o “suelo de ruido” del instrumento. En esta situación, incluso una pequeña cantidad de absorbancia de fondo procedente del tampón o una pequeña desviación del instrumento pueden tener un efecto desproporcionadamente grande en la proporción calculada, haciéndola muy variable y poco fiable. Por lo tanto, las proporciones de pureza de muestras muy diluidas deben considerarse con extremo escepticismo.
En segundo lugar, el pH del tampón utilizado para disolver el ácido nucleico tiene un impacto dramático en la proporción A260/A280. El estado de protonación de las bases nucleotídicas cambia con el pH, lo que a su vez altera su perfil de absorción UV. Medir una muestra de ADN puro en una solución ácida (o en agua no tamponada, que puede ser ligeramente ácida debido al CO2 disuelto) puede reducir la relación A260/A280 hasta en 0,2-0,3 unidades, haciendo que una muestra perfectamente pura parezca contaminada. Por esta razón, la mejor práctica es realizar siempre las mediciones en un tampón estable, bajo en sal y con un pH ligeramente básico, como el tampón TE (Tris-EDTA, pH 8,0), y utilizar exactamente el mismo tampón para la medición del blanco.
Por último, una visión holística de todo el espectro proporciona más información que las proporciones por sí solas. Un único contaminante suele dejar una “huella dactilar” característica en varias longitudes de onda. Por ejemplo, una contaminación importante por proteínas no sólo disminuye la relación A260/A280, sino que también reduce fuertemente la relación A260/A230, porque los enlaces peptídicos de las proteínas absorben fuertemente en el rango de 220-230 nm. Del mismo modo, la contaminación con fenol no sólo disminuye la relación A260/A280, sino que también desplaza característicamente el pico de absorbancia de 260 nm hacia 270 nm. Así pues, el examen del espectro completo puede ayudar a determinar la naturaleza específica de un problema de contaminación.
Tabla 4: Contaminantes comunes en los preparados de ácido nucleico y sus firmas espectroscópicas
| Contaminante | Efecto en A260/A280 | Efecto en A260/A230 | Otras pistas espectrales |
| Proteína | De ligera a moderadamente reducida | Fuertemente reducida | Amplia absorbancia alrededor de 230 nm |
| Fenol | Reducido | Reducido | El pico se desplaza hacia 270 nm. |
| Sales de guanidina | Ligeramente reducido | Fuertemente reducido | Fuerte pico de absorbancia a ~230 nm. |
| EDTA | Sin cambios | Disminuido | El efecto es más pronunciado a bajas concentraciones de ácido nucleico. |
| Etanol | Sin cambios | Ligeramente reducido | No se distingue fácilmente en el espectro. |
| Polisacáridos | Sin cambios | Reducido | Puede aumentar la dispersión de fondo (A320 elevado). |
Parte 3: Cuantificación de proteínas – Medición de las moléculas de la vida
Junto con los ácidos nucleicos, las proteínas son la otra gran clase de biomoléculas cuantificadas rutinariamente por espectrofotometría. Los métodos para la cuantificación de proteínas son más variados, cada uno con distintas ventajas, desventajas y limitaciones críticas que el usuario debe comprender para seleccionar la técnica adecuada.
3.1 El enfoque directo: Absorbancia UV a 280 nm (A280)
El método más sencillo para cuantificar proteínas consiste en medir directamente su absorbancia en el rango UV. Este método se basa en el hecho de que los aminoácidos aromáticos, concretamente el triptófano (Trp) y la tirosina (Tyr), contienen estructuras en anillo que absorben la luz UV con un pico máximo a 280 nm aproximadamente.
Las ventajas del método A280 son convincentes: es extremadamente rápido, no requiere reactivos adicionales y no es destructivo, lo que significa que la muestra puede recuperarse y utilizarse después de la medición. Sin embargo, sus limitaciones son importantes y estrictas.
En primer lugar, el método sólo es adecuado para muestras de proteínas puras. Cualquier sustancia contaminante que también absorba a 280 nm, sobre todo los ácidos nucleicos (que tienen un importante hombro de absorbancia a 280 nm), interferirá en la lectura y dará lugar a una sobreestimación de la concentración de proteínas. En segundo lugar, el método tiene una sensibilidad relativamente baja y requiere una concentración de proteína bastante alta, normalmente superior a 0,1 mg/mL, para generar una señal fiable.28 Por último, y lo más importante, la precisión del método A280 depende del conocimiento del coeficiente de extinción específico de la proteína(ϵ). Este valor depende en gran medida de la composición única de aminoácidos de la proteína, en concreto, de su número de residuos de triptófano y tirosina. Utilizar una fórmula genérica, como la fórmula Warburg, sólo proporciona una estimación aproximada, ya que el coeficiente de extinción puede variar drásticamente de una proteína a otra.
3.2 La revolución colorimétrica: Cuando la medición directa no es suficiente
En el caso de mezclas complejas de proteínas, como los lisados celulares brutos, o cuando la concentración de proteínas es baja, el método A280 directo resulta inadecuado. En estos casos, los investigadores recurren a los ensayos colorimétricos. Se trata de reacciones químicas que producen un producto coloreado en presencia de proteínas. La intensidad del color resultante, que se mide con el espectrofotómetro en el rango de luz visible (por ejemplo, a 562 nm o 595 nm), es proporcional a la concentración de proteína en la muestra.
La principal ventaja de los ensayos colorimétricos es que suelen ser más sensibles que el método A280 y son mucho menos susceptibles a las interferencias de componentes no proteicos que pueda haber en el tampón de la muestra. Sin embargo, todos comparten un requisito crítico: necesitan la creación de una curva estándar. Esto implica ensayar una serie de muestras que contengan una concentración conocida de una proteína estándar (como la albúmina de suero bovino, BSA) junto con las muestras desconocidas. La concentración de la desconocida se determina entonces comparando su absorbancia con esta curva estándar.
3.3 Una inmersión profunda en los ensayos colorimétricos: Un análisis comparativo
Existen varios ensayos colorimétricos, pero la elección entre ellos no es arbitraria. La selección del ensayo más adecuado viene dictada casi siempre por la composición química del tampón en el que se disuelve la muestra de proteína. Ciertos componentes del tampón son incompatibles con determinadas químicas de ensayo, por lo que éste es el factor de decisión más crítico para cualquier investigador.
El ensayo Bradford
- Principio: El ensayo de Bradford se basa en la unión de un colorante, el azul brillante Coomassie G-250, a las proteínas. En su estado no unido, ácido, el colorante es de color marrón rojizo y tiene un máximo de absorbancia a 465 nm. Cuando se une a la proteína, principalmente a través de interacciones con residuos de aminoácidos básicos (por ejemplo, arginina) y aromáticos, el colorante se estabiliza en su forma azul, haciendo que el máximo de absorbancia se desplace a 595 nm.28 El aumento de la absorbancia a 595 nm es proporcional a la cantidad de proteína.
- Ventaja clave: Su química no se ve afectada en gran medida por la presencia de agentes reductores como el ditiotreitol (DTT) y el β-mercaptoetanol, muy comunes en los tampones de proteínas para evitar la oxidación y mantener la estructura de las proteínas.
- Desventaja clave: El ensayo es muy incompatible con detergentes como el dodecil sulfato sódico (SDS) y el Tritón X-100. Estas sustancias químicas interfieren en la interacción colorante-proteína, lo que da lugar a resultados inexactos.
El ensayo BCA (ácido bicinconínico)
- Principio: El ensayo BCA es un método basado en el cobre que implica dos reacciones secuenciales. En primer lugar, en condiciones alcalinas, los enlaces peptídicos de la proteína quelan iones cúpricos (Cu2+) y los reducen a iones cuprosos (Cu+). Esto se conoce como la reacción de biuret. En segundo lugar, dos moléculas de ácido bicinchonínico (BCA) se quelan específicamente con un ion cuproso (Cu+), formando un complejo de color púrpura intenso que presenta un fuerte máximo de absorbancia a 562 nm.
- Ventaja clave: El ensayo BCA es compatible con una amplia gama de detergentes (a menudo hasta una concentración del 5%), lo que lo convierte en el método de elección para muestras solubilizadas a partir de membranas celulares o preparadas en tampones de lisis que contienen detergentes. También presenta menos variabilidad de proteína a proteína en comparación con el ensayo Bradford, ya que la reacción implica la columna vertebral peptídica, que es común a todas las proteínas.
- Desventaja clave: La química del ensayo se basa en la reducción del cobre, por lo que es altamente incompatible con agentes reductores (como el DTT) y agentes quelantes del cobre (como el EDTA), que interfieren directamente con la reacción y conducen a una gran sobreestimación de la concentración de proteínas.
El ensayo de Lowry
- Principio: El ensayo de Lowry es otro método de dos pasos basado en el cobre, históricamente muy importante en bioquímica. Al igual que el ensayo BCA, comienza con la reacción de la proteína con el cobre en una solución alcalina. Sin embargo, el segundo paso implica la reducción del reactivo Folin-Ciocalteu por la proteína tratada con cobre, lo que produce un complejo de color azul.
- Contexto histórico: Durante muchos años, el artículo de Lowry fue uno de los más citados de toda la literatura científica. Sin embargo, el método es más complejo de realizar, sus reactivos son menos estables y es propenso a la interferencia de una gama más amplia de sustancias que el ensayo BCA. Por estas razones, ha sido sustituido en gran medida en los laboratorios modernos por el método BCA, más robusto y cómodo.
Tabla 5: Comparación directa de los ensayos habituales de cuantificación de proteínas
| Método de ensayo | Principio | Longitud de onda (nm) | Compatible con… | Incompatible con… | Pros | Contras |
| UV A280 | Absorbancia UV directa por aminoácidos aromáticos | 280 | La mayoría de los tampones | Ácidos nucleicos, otros compuestos absorbentes de UV | Muy rápido, no destructivo, sin reactivos | Baja sensibilidad, requiere proteína pura, alta variabilidad entre proteínas |
| Bradford | Fijación del colorante Coomassie | 595 | Agentes reductores (DTT), la mayoría de las sales | Detergentes (SDS, Triton X-100), tampones básicos | Rápido, sencillo, alta sensibilidad | Alta variabilidad entre proteínas, incompatibilidad con detergentes |
| BCA | Reducción y quelación del cobre por BCA | 562 | Detergentes (hasta el 5%), la mayoría de tampones | Agentes reductores (DTT), quelantes (EDTA) | Baja variabilidad proteína a proteína, compatible con detergentes | Más lento (requiere incubación), incompatibilidad con agentes reductores |
| Lowry | Reducción de cobre y reactivo Folin | 650-750 | Detergentes | Agentes reductores, quelantes, muchas otras sustancias | Alta sensibilidad | Procedimiento complejo, muchas interferencias, sustituido en gran medida por el BCA |
3.4 El arte de la curva estándar: Una guía paso a paso
En cualquier ensayo colorimétrico, el valor de absorbancia de una muestra desconocida carece de significado por sí solo. Su concentración sólo puede determinarse comparando su absorbancia con una curva estándar generada a partir de una serie de muestras con concentraciones conocidas. La creación de una curva estándar precisa es un paso innegociable para una cuantificación fiable.
El proceso es sencillo pero requiere precisión:
- Prepare una solución madre: Comience con una concentración de alta calidad y exactamente conocida de una proteína estándar. Una elección común es la albúmina de suero bovino (BSA) a una concentración de 2 mg/mL.
- Cree diluciones seriadas: A partir de esta solución madre, prepare una serie de diluciones para crear al menos 5-7 estándares con concentraciones conocidas y decrecientes que abarquen el rango esperado de sus muestras desconocidas. El tampón utilizado para la dilución debe ser el mismo que el tampón en el que se encuentran sus muestras desconocidas.
- Estándares de ensayo y muestras desconocidas: Es crítico procesar todos los estándares y muestras desconocidas al mismo tiempo y bajo condiciones idénticas. Añada el reactivo del ensayo a todos los tubos, mezcle e incube durante el tiempo y a la temperatura especificados.
- Mida la absorbancia: Tras la incubación, mida la absorbancia de todos los estándares y muestras desconocidas a la longitud de onda adecuada para el ensayo elegido (por ejemplo, 595 nm para Bradford, 562 nm para BCA). Recuerde primero “poner en blanco” el espectrofotómetro utilizando una muestra que contenga sólo el tampón y el reactivo del ensayo, sin proteínas.
- Trace los datos: Utilizando un programa informático como Microsoft Excel, cree un gráfico de dispersión. Las concentraciones conocidas de los estándares deben estar en el eje X (la variable independiente), y sus correspondientes valores de absorbancia corregidos por el blanco deben estar en el eje Y (la variable dependiente).
- Genere una línea de tendencia y una ecuación: Ajuste una línea de tendencia a los puntos de datos de los estándares. Para la mayoría de los ensayos en su rango lineal, una regresión lineal es apropiada. Para ensayos con un rango no lineal más amplio, un polinomio de segundo orden (u otro ajuste de curva) puede ser más preciso. Muestre la ecuación de la línea (por ejemplo, y=mx+c) y el coeficiente de determinación (R2) en el gráfico. Elvalor R2 de
es una medida de lo bien que la línea se ajusta a los datos; un valor de 0,99 o superior indica un ajuste muy bueno. - Calcule la concentración desconocida: Con la ecuación de la recta, ahora puede determinar la concentración de su muestra desconocida. Sustituya la absorbancia medida de su muestra desconocida por ‘y’ en la ecuación y resuelva para ‘x’ (concentración). Si su muestra desconocida se diluyó antes del ensayo, recuerde multiplicar esta concentración calculada por el factor de dilución para hallar la concentración de su muestra original sin diluir.
3.5 Elección de su estándar: Por qué la BSA no es siempre la respuesta
Una última consideración crucial es la elección del propio estándar proteico. Esta elección es una importante fuente potencial de error y variabilidad en la cuantificación de proteínas.
La cuestión subyacente es que las diferentes proteínas tienen diferentes composiciones de aminoácidos. Dado que los ensayos colorimétricos como Bradford y BCA se basan en reacciones con residuos de aminoácidos específicos (por ejemplo, arginina para Bradford, cisteína/triptófano para BCA), la cantidad de color producida por microgramo de proteína variará de una proteína a otra. Esto significa que si utiliza BSA como estándar, la concentración que calcule para su proteína de interés es en realidad una concentración “equivalente a BSA”. Es una estimación relativa al estándar, no un valor absoluto.
El enfoque de referencia es utilizar una concentración conocida y altamente purificada de su proteína de interés real para generar la curva estándar. Esto proporcionará la medición más precisa. Sin embargo, para muchos investigadores, esto es poco práctico, ya que una versión purificada de su proteína puede no estar disponible o puede ser demasiado preciada o cara para utilizarla como estándar.
Por lo tanto, la práctica más común y aceptada es utilizar un estándar proteico fácilmente disponible y barato como la albúmina de suero bovino (BSA) o la gammaglobulina bovina (BGG). El principio más importante en este contexto es la coherencia. Al utilizar siempre el mismo estándar proteínico para todos los experimentos relacionados, el investigador se asegura de que, aunque los valores absolutos de concentración puedan ser estimaciones, las comparacionesrelativas entre diferentes muestras sean válidas y reproducibles.
Parte 4: Más allá de la pureza – Medición del crecimiento microbiano
Además de cuantificar las moléculas purificadas, los espectrofotómetros son herramientas esenciales en microbiología para controlar el crecimiento de los cultivos celulares.
4.1 Seguimiento de la vida en un tubo de ensayo: La medida OD600
El método estándar para estimar la densidad de un cultivo bacteriano o de levadura en un medio líquido consiste en medir su densidad óptica (DO) a una longitud de onda de 600 nm, una medida comúnmente denominada DO600.
Es fundamental comprender que nose trata de una verdadera medición de la absorbancia. Las células microbianas no están disueltas en la solución, sino que son partículas en suspensión. Estas partículas son mucho más grandes que la longitud de onda de la luz que se está utilizando, por lo que en lugar de absorber la luz, ladispersan. A medida que el haz de luz atraviesa el cultivo, las células desvían la luz en muchas direcciones. El detector del espectrofotómetro, situado directamente en la trayectoria de la luz, mide una disminución de la luz transmitida porque gran parte de ella se ha dispersado y ya no llega al detector. La longitud de onda de 600 nm se elige porque se encuentra en la gama del naranja visible y no es fuertemente absorbida por los componentes comunes de los medios de crecimiento microbiano (como el extracto de levadura o la triptona), lo que minimiza las interferencias de fondo.
4.2 De la densidad óptica al recuento celular: La importancia de la calibración
Dado que una medición de DO600 se basa en la dispersión de la luz, la relación lineal de la ley de Beer-Lambert no se aplica estrictamente, sobre todo a medida que el cultivo se vuelve denso. A densidades celulares elevadas, la luz que es dispersada por una célula puede volver a ser dispersada por otra célula en la trayectoria del detector, un fenómeno conocido como dispersión múltiple. Esto hace que la relación entre la DO600 y la densidad celular real se vuelva no lineal.
Por lo tanto, para obtener un recuento celular preciso (por ejemplo, en células/mL o unidades formadoras de colonias/mL), cada laboratorio debe generar su propia curva de calibración que correlacione las lecturas de DO600 con el número real de células para su cepa microbiana, condiciones de crecimiento y espectrofotómetro específicos. La geometría de la óptica del instrumento (por ejemplo, la distancia de la muestra al detector) afecta a la cantidad de luz dispersa que se capta, por lo que una lectura de OD600 de 0,5 en un instrumento puede no corresponder a la misma densidad celular que una lectura de 0,5 en un modelo diferente.
El proceso de calibración implica:
- Hacer crecer un cultivo del microorganismo de interés.
- Tomar periódicamente muestras del cultivo en crecimiento.
- Para cada muestra, medir inmediatamente su DO600.
- Para exactamente la misma muestra, realizar una serie de diluciones y colocarlas en placas de agar. Tras la incubación, se cuenta el número de colonias para determinar el recuento de células viables, o unidades formadoras de colonias por mililitro (UFC/mL).
- A continuación, se traza un gráfico con la densidad celular real (UFC/mL) en el eje X y la correspondiente medida de DO600 en el eje Y. Esta curva de crecimiento puede utilizarse entonces en futuros experimentos para convertir de forma rápida y fiable una lectura de OD600 en una densidad celular estimada para esa cepa específica y ese conjunto de condiciones.
Parte 5: El arte de la precisión – Una guía práctica para una espectrofotometría impecable
La potencia de un espectrofotómetro sólo es igualada por su sensibilidad al error del usuario. Conseguir resultados precisos y reproducibles no es automático; es el resultado de una técnica cuidadosa y del cumplimiento de las mejores prácticas.
5.1 Las mejores prácticas para obtener resultados fiables
- Calibración y mantenimiento de los instrumentos: Los instrumentos deben calibrarse regularmente según las recomendaciones del fabricante, a menudo utilizando materiales de referencia certificados. Debe dejarse que el instrumento se caliente adecuadamente antes de utilizarlo para garantizar que la potencia de la lámpara sea estable. También es esencial un mantenimiento rutinario, como la limpieza del exterior y del compartimento de muestras.
- Manipulación delas cubetas: Las cubetas son componentes ópticos de precisión y deben tratarse como tales. Utilice siempre cubetas limpias y sin arañazos. Para las mediciones en el rango UV (por debajo de ~340 nm), las cubetas de cuarzo son obligatorias, ya que el plástico y el vidrio absorben la luz UV. Cuando manipule una cubeta, toque únicamente las caras esmeriladas o estriadas; las huellas dactilares en las superficies ópticas transparentes dispersarán y absorberán la luz, provocando lecturas erróneas. Para obtener la máxima precisión, utilice siempre la misma cubeta para el blanco y la muestra, asegurándose de que esté orientada en la misma dirección en el soporte cada vez.
- Blanqueo adecuado: La medición del “blanco” es posiblemente el paso más crítico para la precisión. La finalidad del blanco es poner a cero el instrumento y restar cualquier absorbancia que proceda del propio disolvente o tampón, o de la cubeta. La solución en blanco debe contener todo lo que contiene la solución de muestra, excepto el analito de interés. Por ejemplo, si su ADN está disuelto en tampón TE, el blanco debe ser tampón TE de la misma reserva. Hacer el blanco con agua cuando su muestra está en un tampón es un error común y grave.
- Preparación de la muestra: Las muestras deben ser homogéneas y estar libres de partículas o burbujas. Agite o mezcle suavemente la muestra inmediatamente antes de realizar una medición para asegurarse de que no hay gradientes de concentración. Si la muestra está turbia, debe centrifugarse para pelar cualquier resto antes de la medición. Al pipetear en la cubeta, evite introducir burbujas de aire, que provocarán una dispersión significativa de la luz y lecturas muy inexactas.
- Rango óptimo de absorbancia: Como dicta la ley de Beer-Lambert, las mediciones sólo son fiables dentro del rango lineal del instrumento. Éste suele situarse entre 0,1 y 1,5 unidades de absorbancia. Las muestras demasiado concentradas (A > 1,5) o demasiado diluidas (A < 0,1) darán resultados poco fiables y deberán diluirse o concentrarse en consecuencia.
- Especificaciones del espectrofotómetro de microvolúmenes: Para instrumentos como el NanoDrop que utilizan una gota de muestra de 1-2 µl, es imprescindible limpiar los pedestales de medición superior e inferior con una toallita limpia, seca y sin pelusa antes de cargar el blanco y antes y después de cada medición de muestra. Utilice una punta de pipeta nueva y una alícuota nueva de muestra para cada lectura a fin de evitar la contaminación cruzada y los efectos de la evaporación.
5.2 Una guía completa para la resolución de problemas
Incluso con una técnica cuidadosa, pueden surgir problemas. La siguiente tabla proporciona una guía para diagnosticar y resolver los problemas más comunes que se encuentran durante la espectrofotometría.
Tabla 6: Guía exhaustiva para la resolución de problemas comunes en espectrofotometría
| Síntoma / Problema | Posibles causas | Soluciones recomendadas |
| Lecturas inestables o a la deriva | 1. La lámpara del instrumento no se ha calentado lo suficiente.
2. La muestra está demasiado concentrada (fuera del intervalo lineal). 3. La muestra no está mezclada; la concentración está cambiando en el recorrido de la luz. 4. Cubeta/pedestal sucios o contaminados. 5. Burbujas de aire en la muestra. |
1. Deje que el instrumento se caliente durante al menos 15-30 minutos.
2. Diluya la muestra para que la absorbancia se sitúe en el intervalo de 0,1-1,5. 3. Agite suavemente la muestra antes de la medición. 4. Limpie a fondo las superficies de la cubeta o del pedestal. 5. Retire la cubeta, golpee suavemente para desalojar las burbujas y vuelva a medir. Vuelva a pipetear si es necesario. |
| Lecturas de absorbancia negativas | 1. La solución en blanco estaba más “sucia” o era más absorbente que la muestra.
2. La cubeta/pedestal estaba sucia durante la medición del blanco y limpia durante la medición de la muestra. 3. El blanco y la muestra se midieron en cubetas diferentes, no coincidentes. |
1. Asegúrese de que el blanco tiene exactamente el mismo tampón/disolvente que la muestra.
2. Vuelva a limpiar la cubeta/pedestal, vuelva a poner en blanco el instrumento y vuelva a medir la muestra. 3. Utilice exactamente la misma cubeta para las mediciones del blanco y de la muestra. |
| Picos inesperados en el espectro | 1. Contaminación en la muestra o en el tampón.
2. Cubeta sucia o rayada. 3. Utilización de un tipo de cubeta incorrecto (por ejemplo, de plástico para mediciones UV). |
1. Prepare una muestra nueva o utilice un nuevo lote de tampón.
2. Limpie a fondo la cubeta o utilice una nueva sin rayar. 3. Utilice una cubeta de cuarzo para cualquier medición por debajo de 340 nm. |
| La relación A260/A280 del ácido nucleico es baja (<1,7) | 1. Contaminación por proteínas.
2. Contaminación por fenol residual. 3. Medido en agua o en un tampón ácido. 4. La concentración de la muestra es demasiado baja (<20 ng/µL), lo que hace que la proporción no sea fiable. |
1. Vuelva a purificar la muestra (por ejemplo, realice un paso de limpieza adicional).
2. Realice una precipitación con etanol o una limpieza en columna para eliminar el fenol. 3. Vuelva a suspender la muestra en una solución tamponada (por ejemplo, TE, pH 8,0) y vuelva a medir. 4. Concentre la muestra o utilice un método de cuantificación más sensible (por ejemplo, fluorescencia). |
| La relación ácido nucleico A260/A230 es baja (<2,0) | 1. Contaminación con sales de guanidina del kit de purificación.
2. 2. Contaminación con carbohidratos, fenol u otros compuestos orgánicos. 3. Blanqueo con un tampón incorrecto (por ejemplo, blanqueo con agua para una muestra en TE). |
1. Asegúrese de que los pasos de lavado de la columna se realizaron correctamente; vuelva a purificar si es necesario.
2. Vuelva a purificar la muestra utilizando un método diferente o pasos de limpieza adicionales. 3. Vuelva a limpiar con el tampón correcto y vuelva a medir la muestra. |
| No hay lectura / Error del instrumento | 1. La lámpara se ha fundido o está fallando.
2. El instrumento no está correctamente conectado al ordenador. 3. Problema de software o ajustes incorrectos. |
1. Compruebe el estado de la lámpara en el software del instrumento; puede que sea necesario sustituir la lámpara.
2. Compruebe todas las conexiones de los cables. 3. Reinicie el software y/o el instrumento. Consulte el manual del usuario para conocer los códigos de error específicos. |
Conclusión: El poder perdurable de un simple haz de luz
El espectrofotómetro es un testimonio del poder de los principios físicos fundamentales aplicados a cuestiones biológicas. Su notable versatilidad le permite cuantificar los componentes básicos de la vida -ADN, ARN y proteínas- e incluso seguir el crecimiento de organismos vivos, todo ello mediante la medición elegante y no invasiva de un simple haz de luz. Desde proporcionar una comprobación rápida de la concentración que lleva apenas unos segundos hasta permitir estudios cinéticos complejos que se desarrollan durante horas, su papel en el laboratorio moderno de ciencias de la vida es a la vez fundacional e indispensable.
Sin embargo, esta guía ha subrayado una verdad crítica: la precisión de un espectrofotómetro no es automática. Es el resultado directo de la comprensión por parte del usuario de la teoría subyacente, de una conciencia aguda de los posibles escollos y fuentes de error, y de una adhesión disciplinada a una práctica de laboratorio meticulosa. Al comprender el recorrido de la luz a través del instrumento, el significado que se esconde tras los índices de pureza, las diferencias cruciales entre los ensayos de proteínas y los matices de la medición de la dispersión de la luz frente a la absorbancia, un investigador transforma el espectrofotómetro de una simple máquina en un poderoso socio analítico. Al dominar esta herramienta esencial, garantizamos la integridad de nuestros datos y, por extensión, la validez de los innumerables descubrimientos que ayuda a posibilitar.
Para adquirir espectrofotómetros HINOTEK, consulte nuestro catálogo.
Para cualquier consulta o necesidad de compra, póngase en contacto con el equipo de HINOTEK: [email protected].
El mantenimiento de esta guía corre a cargo del equipo técnico principal de HINOTEK, compuesto por ingenieros superiores y científicos de aplicaciones con más de dos décadas de experiencia práctica en campos como la microscopía, la centrifugación y la espectrofotometría. Nos comprometemos a garantizar que cada dato de esta guía -desde los principios de los instrumentos y las especificaciones técnicas hasta los consejos para la adquisición de equipos de laboratorio- mantenga el máximo nivel de precisión y actualidad.
Este contenido se revisa y actualiza periódicamente para reflejar los últimos estándares de la industria y los avances tecnológicos. Valoramos los comentarios de la comunidad científica mundial. Si tiene alguna pregunta o sugerencia, o desea comentar algún detalle técnico, no dude en ponerse en contacto con nuestro equipo de expertos en [email protected].