Introducción: El reto analítico de cuantificar la cafeína en matrices complejas
Hoy en día, el té con leche, el café y otras bebidas son cada vez más populares en todo el mundo. La cafeína (C8H10N4O2) es un alcaloide purínico de origen natural y la sustancia psicoactiva más consumida en todo el mundo. Se encuentra en las hojas, semillas y frutos de al menos 63 especies de plantas y es un ingrediente clave del café, el té, los refrescos, las bebidas energéticas y muchas fórmulas farmacéuticas. Por lo tanto, no dé por sentado que porque beba té con leche en lugar de café no afectará a su sueño. De hecho, el contenido de cafeína de muchas bebidas es bastante elevado, lo que puede hacer que se quede despierto mirando al techo en mitad de la noche. En realidad, si dispone de un espectrofotómetro UV Visible HINOTEK 752G (con un precio inferior a 1.000 dólares) y algunos disolventes sencillos, puede incluso medir usted mismo en casa el contenido de cafeína de sus bebidas. El espectrofotómetro es un instrumento rápido y rentable para la cuantificación de la cafeína. La relativa sencillez del instrumento y la naturaleza directa de los principios subyacentes lo convierten en un elemento básico en los laboratorios académicos y en los entornos industriales de garantía de calidad/control de calidad (GC/CC).
Sin embargo, la aparente sencillez de la medición espectrofotométrica oculta un importante reto analítico: la complejidad de la matriz de la muestra. Las bebidas no son soluciones puras de cafeína. Son intrincadas mezclas químicas que contienen una amplia variedad de otros compuestos, como azúcares, colorantes naturales o artificiales, ácidos, conservantes y, en el caso del café y el té, otros alcaloides estructuralmente relacionados. Estas sustancias coexistentes, o “interferencias”, también pueden absorber la luz ultravioleta, a menudo en la misma región que la cafeína, lo que da lugar a importantes errores analíticos. En consecuencia, una medición directa de una muestra de bebida es casi siempre inviable. El verdadero reto analítico, por tanto, no reside en la lectura instrumental final, sino en el paso crítico y previo de la preparación de la muestra. La exactitud, precisión y fiabilidad generales de todo el método vienen dictadas fundamentalmente por la eficacia del proceso de extracción utilizado para aislar la cafeína de su compleja matriz. Este artículo proporciona una guía exhaustiva, a nivel de experto, para superar este reto, detallando los principios fundamentales, los protocolos experimentales sólidos y las consideraciones avanzadas necesarias para la determinación espectrofotométrica precisa de la cafeína en las bebidas.
Sección 1: Principios fundamentales de la cuantificación espectrofotométrica
1.1 La física de la absorción de la luz: Espectroscopia UV-Visible
La capacidad de cuantificar la cafeína mediante este método se basa en los principios fundamentales de cómo interactúan las moléculas con la luz. El espectrofotómetro UV-Visible mide la absorción de la luz en las regiones ultravioleta (190-400 nm) y visible (325-1100 nm) del espectro electromagnético.
Cromóforos y sistemas conjugados
Cuando una molécula absorbe un fotón de luz ultravioleta o visible, la energía del fotón promueve un electrón desde un orbital de estado básico de menor energía (S0) a un orbital de estado excitado de mayor energía (S1). La parte específica de una molécula responsable de esta absorción se denomina cromóforo.
La cafeína es un cromóforo excelente porque su estructura molecular contiene un sistema conjugado:una serie de enlaces simples y dobles alternados. Esta conjugación deslocaliza el
π electrones a través de la molécula, lo que disminuye la brecha energética entre los estados básico y excitado. Como resultado, la cafeína puede absorber fotones de menor energía, concretamente en el rango UV, lo que la hace fácilmente detectable por espectrofotometría UV-Vis.
El espectro de absorción y λmax
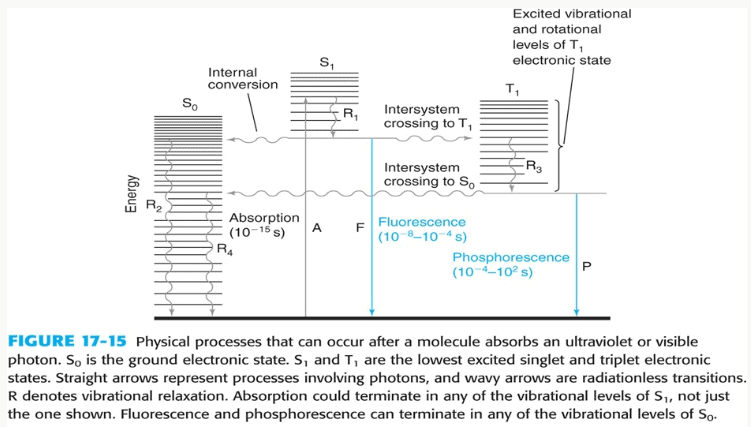 |
Un espectro de absorción es un trazado de la absorbancia de una sustancia frente a la longitud de onda. Para un compuesto determinado en un disolvente específico, este espectro es una huella dactilar única. La longitud de onda a la que se produce la absorbancia máxima se conoce como λmax (lambda max). El análisis cuantitativo se realiza casi siempre en λmaxpor dos razones fundamentales:
- Máxima sensibilidad: En esta longitud de onda, la sustancia absorbe la luz con mayor intensidad, lo que significa que incluso pequeñas concentraciones producen una señal medible. Esto conduce a límites de detección más bajos.
- Precisión y linealidad: La medición de la absorbancia es menos sensible a pequeños errores en la calibración de la longitud de onda en el pico de una banda de absorción amplia, lo que garantiza un mejor cumplimiento de la ley de Beer-Lambert.
Inmersión profunda en la instrumentación
Un espectrofotómetro UV-Vis consta de varios componentes clave que trabajan conjuntamente para producir una medición.
|
|
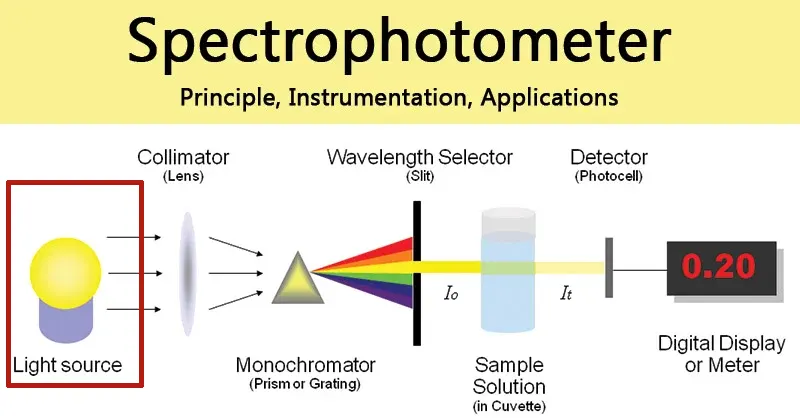
- Fuentes de luz: Para cubrir toda la gama UV-Vis, los instrumentos suelen utilizar dos lámparas. Una lámpara de deuterio proporciona un espectro continuo en la región UV (normalmente 190-350 nm), mientras que una lámpara halógena de tungsteno se utiliza para las regiones visible e infrarroja cercana (normalmente 330 nm y superiores).
- Selección de la longitud de onda (monocromador): Este es el corazón del espectrofotómetro. La luz procedente de la fuente entra en el monocromador a través de una rendija de entrada y se dirige hacia una rejilla de difracción. La rejilla dispersa la luz en sus longitudes de onda constituyentes, de forma muy parecida a un prisma. Al girar la rejilla, se puede seleccionar una banda específica y estrecha de longitudes de onda para que pase a través de una rendija de salida y llegue a la muestra. La anchura de la rendija de salida controla elancho de banda espectral de
, que es una medida de la gama de longitudes de onda que pasan a través de ella. Un ancho de banda más estrecho proporciona una mejor resolución espectral (la capacidad de distinguir entre dos picos cercanos) pero permite que llegue menos luz al detector, lo que aumenta potencialmente el ruido de la señal. - Compartimento de la muestra y cubetas: La luz monocromática pasa a través de la muestra, que se mantiene en un pequeño recipiente transparente llamado cubeta. Para el análisis en la región UV, las cubetas deben ser de cuarzo, ya que el vidrio y el plástico estándar absorben la radiación UV e interferirían en la medición.
- Detectores: Tras atravesar la muestra, la luz transmitida incide en un detector, que convierte la intensidad luminosa en una señal eléctrica medible. Entre los detectores más comunes se encuentran los tubos fotomultiplicadores (PMT), que son muy sensibles y estables, y los fotodiodos de silicio, que son robustos y económicos.
- Diseño óptico: Los espectrofotómetros pueden tener un diseño de haz único o de doble haz. En un instrumento de haz único, el blanco y la muestra deben medirse secuencialmente. Un instrumento de doble haz divide el haz de luz, pasando uno a través de la muestra y el otro a través de una referencia (blanco) simultáneamente. Este diseño corrige automáticamente las fluctuaciones de la intensidad de la lámpara a lo largo del tiempo, proporcionando una línea de base más estable y una mayor precisión.
1.2 La ley de Beer-Lambert: El eje del análisis cuantitativo
La ley de Beer-Lambert (a menudo abreviada como ley de Beer) es el fundamento matemático que relaciona la cantidad de luz absorbida por una muestra con la concentración del analito que contiene. La ley se expresa como A=ϵbc
Donde cada término tiene un significado preciso :
- A (Absorbancia): Cantidad adimensional que representa la cantidad de luz absorbida por la muestra. Se define en una escala logarítmica como A=log10(I0/I), donde I0 es la intensidad de la luz incidente (medida con una solución “en blanco” que sólo contiene el disolvente) e I es la intensidad de la luz transmitida a través de la muestra. La relación logarítmica es crucial porque crea una correlación lineal entre la absorbancia y la concentración, con la que es mucho más fácil trabajar que con la relación exponencial de la transmitancia.
- ϵ (Absortividad molar): También conocido como coeficiente de extinción molar, es una constante característica de una sustancia específica a una longitud de onda concreta (λmax). Representa la intensidad con la que la sustancia absorbe la luz. Sus unidades suelen ser litros por centímetro molar (L⋅mol-1⋅cm-1). Una alta absortividad molar indica una alta probabilidad de la transición electrónica, lo que significa que la sustancia es un fuerte absorbente de luz en esa longitud de onda.
- b (Longitud del camino): Es la distancia que recorre la luz a través de la muestra, que viene determinada por la anchura interna de la cubeta. Se estandariza casi universalmente a 1 cm para facilitar la comparación de resultados entre instrumentos y laboratorios.
- c (Concentración): Es la concentración molar de la sustancia absorbente en la solución (mol⋅L-1), que es la cantidad que se desea determinar.
La ley de Beer-Lambert establece que, para una sustancia dada a una longitud de onda y un recorrido fijos, la absorbancia es directamente proporcional a la concentración. Esta relación lineal es la que permite crear una curva de calibración, un gráfico de la absorbancia frente a una serie de concentraciones conocidas, que puede utilizarse después para determinar la concentración de una muestra desconocida.
Si desea más información sobre el espectrofotómetro, consulte nuestra página: ¿Qué es un espectrofotómetro y cómo funciona? La guía definitiva
Sección 2: Un protocolo paso a paso para el análisis de la cafeína
Esta sección ofrece un flujo de trabajo detallado y práctico para la determinación cuantitativa de la cafeína. El éxito depende de una técnica meticulosa, especialmente en la preparación de los estándares y la extracción de la muestra.
2.1 Preparación de las soluciones patrón y construcción de la curva de calibración
La curva de calibración es la referencia con respecto a la cual se mide la muestra desconocida. Por tanto, su precisión es primordial.
Reactivos y material de vidrio
Todo el trabajo debe realizarse utilizando reactivos y equipos de grado analítico para minimizar los errores. Esto incluye
- Un estándar de cafeína de alta pureza (≥99%).
- Disolventes de grado analítico (por ejemplo, diclorometano, cloroformo o agua desionizada).
- Matraces aforados y pipetasde clase A para la preparación precisa de las soluciones.
Preparación de la solución madre
Una solución madre concentrada sirve como fuente para todos los estándares posteriores.
- Utilice una balanza analítica para pesar con exactitud una masa precisa del estándar de cafeína pura (por ejemplo, 100,0 mg).
- Transfiera cuantitativamente el sólido a un matraz aforado del tamaño adecuado (por ejemplo, 100,0 mL).
- Añada una porción del disolvente elegido, agite para disolver completamente el sólido y, a continuación, diluya hasta la marca de calibración con el disolvente. Tape e invierta el matraz varias veces para garantizar la homogeneidad.
Este procedimiento crearía una solución madre de 1000 mg/L (o 1000 ppm).
Preparación de estándares de trabajo mediante dilución en serie
A partir de la solución madre se prepara una serie de 4 a 6 estándares de concentración decreciente. Por ejemplo, para preparar un patrón de 10 ppm a partir de una solución madre de 100 ppm en un matraz aforado de 50 mL, se utilizaría la ecuación de dilución M1V1=M2V2:
(100 ppm)×V1=(10 ppm)×(50 mL), lo que da V1=5 mL.
Así, se pipetearían 5,00 mL de la solución madre en el matraz de 50 mL y se diluirían hasta la marca. Este proceso se repite para crear una serie de estándares que pongan entre paréntesis la concentración esperada de la muestra desconocida (por ejemplo, 2, 5, 10, 15, 20 ppm).
Determinación experimental de λmax
Aunque la bibliografía suele proporcionar un valor λmax para la cafeína, este valor puede variar en función del disolvente y del pH. Un estudio de los métodos muestra valores comunicados que oscilan entre 260 nm y 276 nm. Adoptar ciegamente un valor bibliográfico sin verificarlo para las condiciones experimentales específicas es una fuente de error común pero significativa, ya que compromete la sensibilidad y la linealidad. Por lo tanto, es una buena práctica crítica determinar λmaxexperimentalmente.
- Seleccione un estándar de cafeína de rango medio (por ejemplo, 10 ppm).
- Con el espectrofotómetro, realice un barrido de longitudes de onda con este patrón en el intervalo UV (por ejemplo, de 190 nm a 350 nm), utilizando el disolvente puro como blanco.
- El software del instrumento generará un espectro. Identifique la longitud de onda correspondiente al pico de absorbancia más alto. Esta es la λmax experimental que debe utilizarse para todas las mediciones posteriores.
Generación y validación de la curva de calibración
- Ajuste el espectrofotómetro a la λmax determinada experimentalmente.
- Ponga a cero el instrumento utilizando una cubeta llena con el disolvente puro (el blanco).
- Mida la absorbancia de cada uno de los estándares de trabajo preparados, empezando por el menos concentrado y avanzando hacia el más concentrado. Enjuague la cubeta con el siguiente estándar antes de llenarla para evitar la contaminación cruzada.
- Trace la absorbancia medida (eje y) frente a la concentración conocida (eje x).
- Realice una regresión lineal sobre los puntos de datos. La salida resultante proporcionará la ecuación de la línea (y=mx+b) y el coeficiente de determinación (R2). Para una calibración válida, elvalor R2 de
debe ser muy cercano a 1 (típicamente >0,995), lo que indica una fuerte relación lineal, y la intersección y (b) debe ser muy cercana a cero.
Tabla 1: Ejemplo de datos de calibración y resultado de la regresión lineal
Esta tabla proporciona un punto de referencia para una calibración satisfactoria utilizando datos hipotéticos.
| Concentración de cafeína (ppm) | Absorbancia medida a 273 nm |
| 0 (blanco) | 0.000 |
| 2.0 | 0.132 |
| 4.0 | 0.259 |
| 8.0 | 0.525 |
| 12.0 | 0.781 |
| 16.0 | 1.035 |
- Gráfico de regresión lineal: Gráfico que muestra estos puntos formando una línea recta.
- Ecuación resultante: Absorbance=0.0645×[Caffeine, ppm]+0.0021
- Coeficiente de determinación: R2=0.9998
2.2 Preparación de la muestra: El paso crítico de la extracción
Se trata de la parte más crucial del análisis, destinada a aislar la cafeína de los componentes de la matriz que interfieren. La elección del método depende del tipo de muestra, del equipo disponible y del rendimiento deseado.
Pretratamiento de la muestra
- Bebidas carbonatadas: Deben desgasificarse a fondo antes de la extracción. Esto puede hacerse agitando con un agitador magnético durante 20 minutos, sonicando o simplemente vertiendo la bebida de un lado a otro entre dos vasos de precipitados hasta que deje de burbujear. De este modo se evita una peligrosa acumulación de presión en el embudo de decantación durante la extracción.
- Muestras sólidas (té/café): Es necesaria una extracción inicial con agua caliente. Se infusiona una masa conocida de hojas de té o posos de café en un volumen conocido de agua hirviendo durante un tiempo determinado (por ejemplo, 20 minutos) para disolver la cafeína en una solución acuosa, que luego se filtra.
Método A: Extracción líquido-líquido (LLE)
La LLE es una técnica clásica basada en la partición de un soluto entre dos fases líquidas inmiscibles.
- Principio: La cafeína es moderadamente soluble en agua pero mucho más soluble en ciertos disolventes orgánicos como el diclorometano (CH2Cl2) o el cloroformo (CHCl3). Cuando la muestra de bebida acuosa se mezcla con uno de estos disolventes, la cafeína pasará preferentemente de la fase acuosa a la orgánica. La eficacia de esta transferencia se describe mediante el coeficiente de partición,k.
- Ajuste del pH: Este es un paso vital para muestras como el té y el café, que contienen compuestos ácidos llamados taninos. Los taninos también son solubles en disolventes orgánicos e interferirán en el análisis. Al añadir una base como el carbonato sódico (Na2CO3) o el hidróxido sódico (NaOH) a la muestra acuosa, los taninos se desprotonan, formando sales iónicas. Estas sales son muy solubles en agua pero insolubles en el disolvente orgánico, por lo que permanecen en la capa acuosa mientras se extrae la molécula neutra de cafeína.
- Procedimiento:
- Coloque un volumen conocido de la bebida desgasificada (por ejemplo, 50 ml) en un embudo de decantación.
- Añada la base (por ejemplo, 1-2 g de Na2CO3 o unos mL de NaOH 1 M) y agite para disolver.
- Añada una porción del disolvente orgánico (por ejemplo, 25 mL de CH2Cl2).
- Tape el embudo e inviértalo suavemente varias veces para mezclar las capas, ventilando con frecuencia abriendo la llave de paso para liberar la presión acumulada. Una agitación enérgica puede crear una emulsión difícil de separar.
- Deje reposar el embudo hasta que las dos capas se hayan separado por completo. El CH2Cl2 y el CHCl3 son más densos que el agua y formarán la capa inferior.
- Vacíe la capa orgánica inferior en un matraz Erlenmeyer limpio.
- Repita la extracción dos veces más con porciones frescas del disolvente orgánico. Las extracciones múltiples con volúmenes más pequeños son significativamente más eficaces para recuperar el analito que una sola extracción con un gran volumen.
- Combine todos los extractos orgánicos.
- Secado y preparación final: El extracto orgánico combinado puede contener trazas de agua disuelta, lo que puede interferir en la medición. Añada una pequeña cantidad de un agente secante anhidro, como sulfato sódico anhidro (Na2SO4), y agite hasta que el líquido sea transparente. Filtre o decante el disolvente seco en un nuevo matraz. Evapore suavemente el disolvente (por ejemplo, utilizando un evaporador rotatorio o un baño de agua caliente en una campana extractora) para dejar el residuo de cafeína en bruto. A continuación, este residuo se disuelve en un volumen preciso y conocido del disolvente de análisis (el mismo utilizado para los estándares) para la medición espectrofotométrica.
Método B: Extracción en fase sólida (SPE)
La SPE es una alternativa más moderna y a menudo más eficaz que la LLE.
- Principio: La SPE utiliza un pequeño cartucho relleno de un material adsorbente sólido (la fase estacionaria), como la sílice C18. La muestra se hace pasar por el cartucho y, en función de la afinidad química, algunos compuestos quedan retenidos en la fase sólida mientras que otros la atraviesan. A continuación, los compuestos retenidos pueden lavarse selectivamente con un disolvente diferente.
- Resumen del procedimiento:
- Acondicionamiento: El cartucho se prepara haciendo pasar por él un pequeño volumen de metanol, seguido de agua desionizada. Esto activa la fase estacionaria C18.
- Carga: La muestra de bebida preparada (a menudo diluida) se hace pasar lentamente a través del cartucho. Las moléculas de cafeína no polares quedan fuertemente retenidas por el sorbente C18 no polar, mientras que las interferencias altamente polares, como los azúcares y las sales, pasan al residuo.
- Lavado: El cartucho se enjuaga con un disolvente débil (por ejemplo, agua) para lavar cualquier resto de interferencias polares débilmente unidas.
- Elución: Se hace pasar por el cartucho un disolvente orgánico fuerte, como el metanol. Este disolvente interrumpe la interacción entre la cafeína y el sorbente C18, lavando (eluyendo) la cafeína purificada en un tubo de recogida limpio.
- Ventajas: En comparación con la LLE, la SPE suele ser más rápida, utiliza mucho menos disolvente, es más fácil de automatizar y a menudo produce un extracto más limpio con tasas de recuperación más altas y reproducibles.
2.3 Medición y cálculo del contenido de cafeína
Medición de la absorbancia
El extracto final de la muestra preparada se transfiere a una cubeta de cuarzo y se mide su absorbancia en el λmax previamente determinado. Es crucial que el valor de absorbancia medido se encuentre dentro del intervalo lineal de la curva de calibración. Un intervalo ideal de absorbancia suele estar entre 0,1 y 1,0. Si la absorbancia es demasiado alta (es decir, por encima del estándar más concentrado), la solución de muestra debe diluirse con precisión con el disolvente y volver a medirse. Esta nueva dilución debe tenerse en cuenta en el cálculo final.
Cálculo de la concentración a partir de la curva de calibración
- Tome la ecuación de regresión lineal obtenida a partir de la curva de calibración: y=mx+b.
- Reorganice la ecuación para resolver la concentración (x):
x=my-b - Sustituya y en la ecuación por la absorbancia medida de la muestra desconocida. El resultado, x, es la concentración de cafeína en la solución final que se midió en la cubeta.
Aplicación del factor de dilución
Este es el último paso crítico. La lectura de la curva se aplica al extracto diluido, no a la bebida en sí. Para obtener la concentración original, multiplique el valor medido por el factor de dilución total.
Concentración (original) = Concentración (medida) × Factor de dilución
Factor de dilución = Volumen final ÷ Volumen inicial
Ejemplo:
- Extraiga 10 mL de bebida y lleve el residuo a 50 mL.
Factor de dilución = 50 ÷ 10 = 5. - Si a continuación diluye esa solución de 50 mL 1 a 10 antes de realizar la prueba, la dilución total pasa a ser 5 × 10 = 50.
Conversión de unidades e informe final
La concentración suele calcularse en mg/L (ppm). Puede que sea necesario convertirlo a una unidad más fácil de usar para el consumidor, como mg por ración. Por ejemplo, para hallar la cantidad en una lata de 355 ml (12 oz):
mg por ración=Concentración (mg/L)×0,355 L/ración
Tabla 2: Ejemplo de cálculo para una bebida hipotética
Esta tabla ilustra el flujo de trabajo completo del cálculo.
| Parámetro | Valor / Paso |
| Datos dados | |
| Ecuación de calibración | Absorbance=0.0645×[Caffeine, ppm]+0.0021 |
| Absorbancia medida de la muestra | 0.450 |
| Volumen inicial de la bebida | 20,0 ml |
| Volumen final de extracto | 100,0 ml |
| Tamaño de la ración de bebida | 355 mL |
| Paso 1: Calcular la concentración en la cubeta | x=(y-b)/m x=(0,450-0,0021)/0,0645 Concentración (medida) = 6,94 ppm (o 6,94 mg/L) |
| Paso 2: Calcule el factor de dilución | Factor dedilución = Volumen final / Volumen inicial Factor de dilución = 100,0 mL / 20,0 mL Factor de dilución = 5 |
| Paso 3: Calcule la concentración en la bebida original | Conc. (original) = Conc. (medida) × Factor de dilución Conc. (original) = 6,94 mg/L × 5 Concentración (original) = 34,7 mg/L |
| Paso 4: Convertir a mg por ración | mg/servida = Conc. (original) × Volumen de la ración (L) mg/servida = 34,7 mg/L × 0,355 L Valor final notificado = 12,3 mg por ración de 355 ml |
Sección 3: Consideraciones avanzadas: Interferencias, limitaciones y solidez del método
Aunque el protocolo descrito es robusto, un analista experto debe ser consciente de sus limitaciones inherentes y de las posibles fuentes de error para generar datos verdaderamente fiables.
3.1 Navegar por el laberinto químico: Interferencias potenciales
- Amplios efectos de la matriz: Incluso con la extracción, pueden arrastrarse al extracto final trazas de compuestos muy coloreados (como el color caramelo de las colas) u otras sustancias que absorben la radiación UV, contribuyendo a un error positivo (una lectura artificialmente alta). Esto se conoce como efecto matriz.
- Solapamiento espectral específico: El problema de la metilxantina: Representa la principal limitación química del método, o su “talón de Aquiles”. La cafeína forma parte de la familia de las metilxantinas, que también incluye la teobromina y la teofilina. Estas moléculas son estructuralmente muy similares a la cafeína y, como resultado, tienen cromóforos similares que absorben la luz UV en longitudes de onda casi idénticas.
- En las bebidas en las que la cafeína es simplemente un aditivo (por ejemplo, la mayoría de las colas), esto no supone un problema importante.
- Sin embargo, en los productos derivados de fuentes naturales como el té, el café y, especialmente, el cacao (utilizado en algunas bebidas energéticas y cafés especiales), la teobromina y la teofilina están presentes de forma natural.
- Una extracción líquido-líquido estándar coextraerá estos alcaloides relacionados junto con la cafeína. Dado que sus espectros de absorción se solapan significativamente con el de la cafeína, el espectrofotómetro no puede distinguirlos. La medición de absorbancia resultante es una suma de la absorbancia de los tres compuestos, lo que conduce a una sobreestimación significativa del contenido real de cafeína. Esta limitación significa que, en el caso de los productos naturales complejos, el método UV-Vis simple se utiliza mejor como herramienta de cribado, y sus resultados deben interpretarse con precaución.
3.2 Estrategias para mejorar la precisión y la especificidad
Para matrices en las que las interferencias son un problema conocido, pueden emplearse varias técnicas avanzadas para mejorar la calidad de los resultados.
- Corrección del fondo: A veces puede hacerse una corrección sencilla midiendo la absorbancia en una segunda longitud de onda en la que se sabe que la cafeína no absorbe, pero en la que sí podría hacerlo una interferencia de fondo constante (por ejemplo, 310 nm o 350 nm). Este valor de absorbancia de fondo se resta entonces de la absorbancia medida en λmax.
- Espectrofotometría derivativa: Se trata de una potente técnica matemática que puede resolver bandas espectrales superpuestas sin separación física. El software del instrumento calcula la primera, segunda o incluso tercera derivada del espectro de absorbancia (dA/dλ, d2A/dλ2, etc.). Este proceso puede amplificar las pequeñas diferencias entre los espectros de la cafeína y los interferentes, creando a menudo puntos de “cruce por cero” para un compuesto que pueden utilizarse para cuantificar el otro, o creando picos y valles únicos para cada compuesto que pueden medirse pico a pico. Se trata de una estrategia clave para mejorar la precisión de la determinación de la cafeína en muestras complejas como el café y el té utilizando únicamente un espectrofotómetro.
- Comparación con un método separativo (HPLC): El “patrón oro” para el análisis de la cafeína es la cromatografía líquida de alto rendimiento (HPLC). La HPLC utiliza una bomba de alta presión para hacer pasar la muestra a través de una columna empaquetada con una fase estacionaria. Los componentes de la mezcla interactúan de forma diferente con la columna y se separan físicamente, eluyendo en tiempos diferentes. A continuación, un detector (a menudo un detector UV) cuantifica cada componente a medida que sale de la columna. Dado que proporciona una separación física, la HPLC no es susceptible a los problemas de solapamiento espectral que afectan a la espectrofotometría directa. Para fines de investigación, reglamentarios o de desarrollo de productos, la validación del método UV-Vis frente a la HPLC es esencial para confirmar su exactitud.
3.3 Comprender los límites: Limitaciones de la ley de Beer-Lambert
La relación lineal descrita por la ley de Beer-Lambert sólo es válida en determinadas condiciones ideales. Las desviaciones de esta ley pueden dar lugar a resultados inexactos, especialmente si se extrapola la curva de calibración.
- Desviaciones químicas: La ley supone que las moléculas absorbentes no interactúan entre sí. En concentraciones elevadas (normalmente >0,01 M), las moléculas de soluto pueden acercarse lo suficiente como para afectar a las nubes de electrones de las demás, lo que puede alterar ligeramente su capacidad de absorber luz (absortividad molar). Esto suele hacer que la curva de calibración se curve hacia abajo a concentraciones elevadas, lo que provoca una desviación negativa de la linealidad.
- Desviaciones instrumentales:
Radiación policromática: La ley sólo es estrictamente válida para la luz puramente monocromática (luz de una sola longitud de onda). Los monocromadores del mundo real pasan una banda estrecha de longitudes de onda. Si la absortividad molar del analito cambia significativamente a lo largo de esta banda, pueden producirse desviaciones de la linealidad. Esta es otra razón por la que se prefiere medir en el pico plano de λmax.
Luz parásita: Se refiere a cualquier luz extraña que llegue al detector sin haber atravesado la muestra correctamente (por ejemplo, por reflexiones internas o fugas de luz). La luz parásita se vuelve especialmente problemática a altas absorbancias. A medida que la luz transmitida verdadera (I) se vuelve muy baja, la señal de luz parásita constante se convierte en una fracción significativa de la luz total que incide en el detector, provocando que la absorbancia medida sea artificialmente baja. Esto establece efectivamente un límite superior en el rango de absorbancia útil del instrumento. Los sistemas de monocromador doble están diseñados específicamente para minimizar la luz parásita y ampliar el rango lineal.
Desviaciones físicas (dispersión): Si la solución de la muestra está turbia o contiene partículas en suspensión (por ejemplo, de una filtración incompleta), estas partículas dispersarán el haz de luz. Esta dispersión desvía la luz lejos del detector, lo que el instrumento interpreta como absorción, dando lugar a una lectura de absorbancia falsamente alta. Todas las muestras deben ser visualmente claras y estar libres de partículas.
Conclusión y recomendaciones
La espectrofotometría UV-Visible ofrece un método rápido, rentable y potente para la cuantificación de la cafeína en las bebidas. Su accesibilidad la convierte en una herramienta inestimable para fines educativos y para el control de calidad rutinario en entornos industriales. Este artículo ha demostrado que, si bien la medición instrumental es sencilla, el éxito del método depende críticamente de un protocolo de preparación de la muestra bien diseñado y meticulosamente ejecutado. El principal obstáculo analítico no es la medición de la cafeína en sí, sino su aislamiento eficaz a partir de una matriz química compleja.
A partir del análisis exhaustivo de los principios y los posibles escollos, se ofrecen las siguientes recomendaciones:
- Para matrices simples (por ejemplo, refrescos): La extracción líquido-líquido (LLE) o la extracción en fase sólida (SPE) descritas, seguidas de una medición UV-Vis directa, constituyen un método muy fiable y adecuado. En estas muestras, la cafeína suele ser un aditivo, y la concentración de interferentes que se solapan espectralmente es insignificante.
- Para matrices complejas (por ejemplo, té, café, bebidas que contienen cacao): El analista debe proceder con una mayor conciencia de las interferencias potenciales, principalmente de la coextracción de otras metilxantinas como la teobromina y la teofilina. Aunque un protocolo LLE-UV-Vis estándar puede proporcionar una estimación razonable, es probable que los resultados estén sesgados positivamente. Para mejorar la precisión, se recomienda encarecidamente el uso de la espectrofotometría derivativa, ya que puede resolver matemáticamente los espectros superpuestos y proporcionar una cuantificación más específica.
- Para investigación, desarrollo de métodos y cumplimiento de la normativa: Para las aplicaciones que exigen el mayor grado de precisión, especificidad y defendibilidad legal, el método espectrofotométrico UV-Vis debe considerarse una herramienta de cribado. Los valores finales notificables deben obtenerse o confirmarse mediante una técnica de separación validada , siendo la HPLC el estándar de la industria y la investigación.
En última instancia, la generación de datos analíticos fiables requiere algo más que seguir un procedimiento; exige un conocimiento profundo de la química del método, sus puntos fuertes inherentes y sus limitaciones críticas. Aplicando los principios y protocolos detallados en este artículo, los analistas pueden emplear con confianza la espectrofotometría UV-Vis para producir datos precisos y significativos sobre el contenido de cafeína de las bebidas.
Si está listo para encontrar el espectrofotómetro UV-Vis adecuado para su laboratorio, consulte nuestra gama completa de productos: Espectrofotómetros UV-Visible
Si tiene alguna pregunta, póngase en contacto con nosotros por correo electrónico: [email protected]
El mantenimiento de esta guía corre a cargo del equipo técnico principal de HINOTEK, formado por ingenieros superiores y científicos de aplicaciones con más de dos décadas de experiencia práctica en campos como la microscopía, la centrifugación y la espectrofotometría. Nos comprometemos a garantizar que cada parte de la información contenida en esta guía -desde los principios de los instrumentos y las especificaciones técnicas hasta los consejos para la adquisición de equipos de laboratorio- mantenga el máximo nivel de precisión y actualidad.Este contenido se revisa y actualiza periódicamente para reflejar los últimos estándares de la industria y los avances tecnológicos. Valoramos los comentarios de la comunidad científica mundial. Si tiene alguna pregunta o sugerencia, o desea comentar algún detalle técnico, no dude en ponerse en contacto con nuestro equipo de expertos en [email protected].