The Principle of PCR: A Molecular Photocopier for DNA
|
|

The Polymerase Chain Reaction, or PCR, is a foundational laboratory technique used for the enzymatic amplification of a specific segment of DNA. In essence, it functions as a “molecular photocopier,” capable of generating thousands to millions of identical copies of a particular DNA sequence from an exceptionally small starting sample. This ability to amplify a single DNA molecule across several orders of magnitude makes it possible to detect, analyze, and manipulate genetic material that would otherwise be present in quantities too small to work with.
The power of PCR lies in its core concept of exponential amplification. The process is a chain reaction where the DNA synthesized in each cycle serves as a template for the subsequent cycle. This creates a doubling effect, where the number of copies of the target DNA, often referred to as the amplicon, increases exponentially. The number of copies produced after a given number of cycles, denoted by ‘n’, can be calculated using the formula ‘2n’. After approximately 30 cycles, this process can amplify the target sequence by a factor of one million to one billion, resulting in a sufficient quantity of DNA for downstream analysis, such as visualization on an agarose gel.
Developed by Kary Mullis in the 1980s, PCR was a breakthrough that fundamentally reshaped the landscape of molecular biology. However, the earliest iterations of the technique were far from the streamlined, automated process used today. The original method was a laborious manual procedure that required researchers to physically move samples between three separate water baths set to the precise temperatures needed for each step of the reaction. Furthermore, the DNA polymerase enzyme available at the time, the Klenow fragment of coli DNA polymerase I, was not heat-stable. It was denatured and destroyed during the high-temperature step of each cycle, forcing researchers to add a fresh aliquot of the enzyme after every single cycle.
The critical innovation that transformed PCR from a cumbersome academic exercise into a ubiquitous and indispensable laboratory tool was the discovery and application of a thermostable DNA polymerase. The isolation of Taq polymerase from the thermophilic bacterium Thermus aquaticus, which thrives in high-temperature environments, provided an enzyme that could withstand the repeated heating and cooling cycles of PCR without losing its activity. This single biochemical property was the linchpin that unlocked the potential for automation. Because the enzyme no longer needed to be manually replenished, it became possible to engineer a single, self-contained instrument—the thermal cycler—that could be programmed to perform the entire series of temperature changes automatically. This synergy between a biological discovery and an engineering solution is what enabled the widespread adoption of PCR and cemented its role as a cornerstone of modern life sciences.
The Engine of Amplification: How a PCR Cycle Works
The vast majority of PCR methods are driven by thermal cycling, a process that subjects the reaction mixture to a series of 20 to 40 repeated temperature changes. This precisely controlled sequence of heating and cooling is performed in an instrument called a thermal cycler, which automates the entire process according to a pre-programmed protocol. A typical PCR program begins with an initial, extended heating step to ensure all template DNA is fully separated before the main cycles commence. The core of the reaction then proceeds through three distinct stages, repeated in each cycle.
Stage 1: Denaturation – Separating the DNA Strands
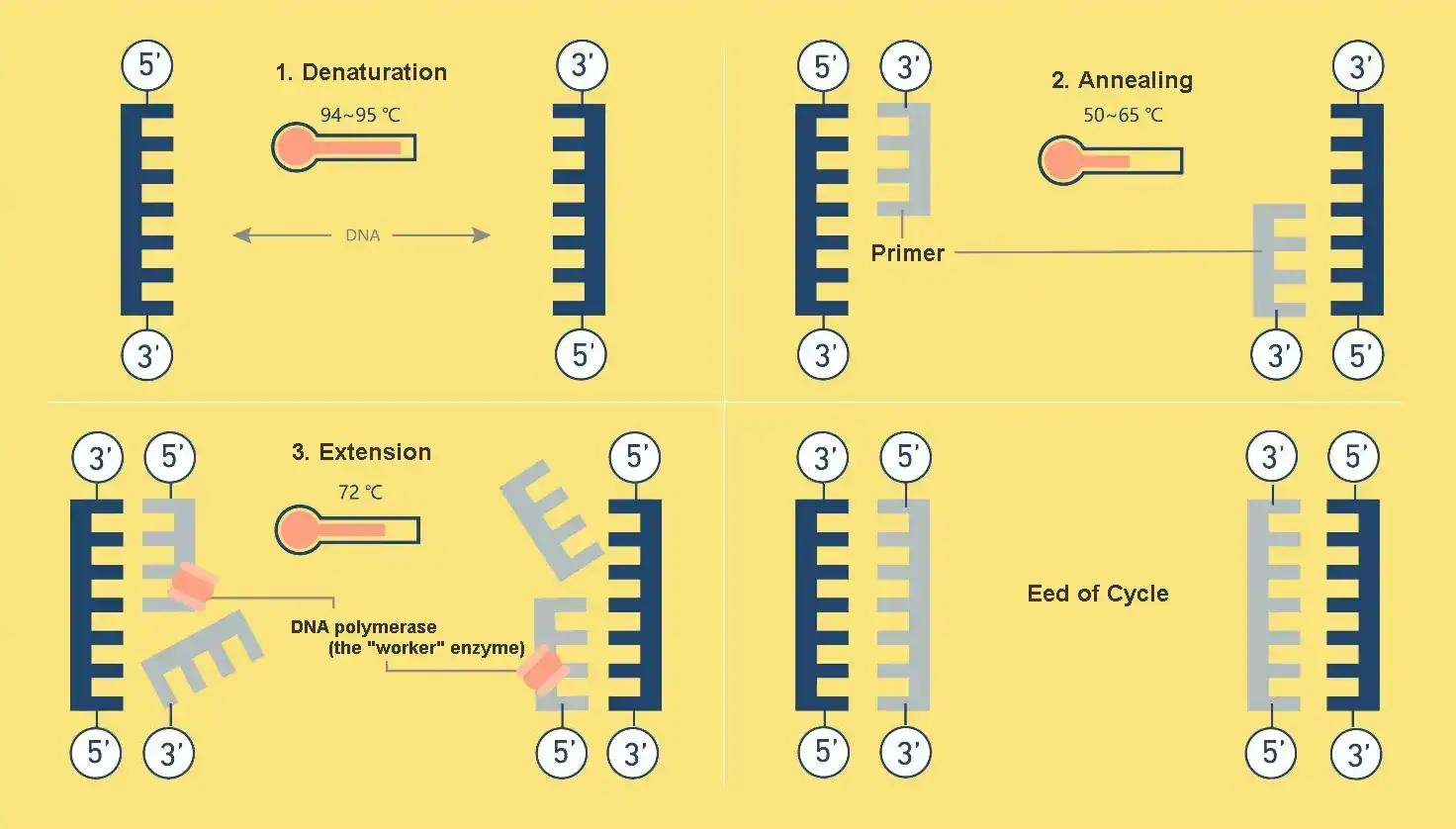 |
The first stage of each cycle is denaturation. The reaction mixture is heated to a high temperature, typically between 94°C and 98°C, for about 15 to 30 seconds. This intense heat provides the energy needed to break the hydrogen bonds that hold the two strands of the DNA double helix together. As these bonds are disrupted, the double-stranded DNA “melts” and separates into two single strands. This step is essential, as it makes the nucleotide sequences of the DNA accessible and provides the single-stranded templates required for the primers to bind in the next stage.
Stage 2: Annealing – Primers Find Their Target
Components and Their Sources in the PCR Reaction Tube at This Stage:
- Single-stranded DNA template: Derived from the original DNA sample you added (e.g., genomic DNA, plasmid DNA, etc.), and produced during the previous high-temperature denaturation step.
- Short single-stranded DNA primers: These are artificially synthesized and pre-added to the reaction tube before the experiment begins. They function like a special piece of “double-sided tape.” One side (its base sequence) firmly adheres (binds) to a specific location on the single-stranded DNA template. The other side (its 3′-OH end) provides an attachment point for the DNA polymerase (the “construction worker”) to start “applying” the new DNA “cement.”
- DNA polymerase: An enzyme (the most commonly used is the heat-stable Taq polymerase), which is also pre-added. It is the “construction worker” here.
- Deoxynucleoside triphosphates (dNTPs): These are the “building materials” for synthesizing the new DNA strand and are also pre-added.
Following denaturation, the temperature of the reaction is lowered significantly, usually to a range between 50°C and 65°C. This reduction in temperature allows the short, single-stranded DNA primers present in the mixture to move and bind, or anneal, to their specific complementary sequences on the single-stranded DNA templates. This step, which typically lasts for 15 to 30 seconds, is the most critical for the success of the reaction, as the annealing temperature directly dictates the specificity of the amplification.
The success of a PCR experiment hinges on a delicate thermodynamic balancing act performed during this annealing step. The chosen annealing temperature (Ta) represents a direct trade-off between the yield of the desired product and the specificity of the reaction. If the temperature is set too low, primers may bind non-specifically to sequences that are only partially complementary, leading to the amplification of incorrect DNA fragments. Conversely, if the temperature is too high, the primers may not bind efficiently enough to the intended target sequence, resulting in a low yield or a complete failure of the reaction. The optimal annealing temperature is therefore not a fixed value but a narrow window, typically 3–5°C below the calculated melting temperature (Tm) of the primers, which maximizes specific binding while minimizing off-target events. This challenge of optimization explains the value of specific equipment features, such as gradient thermal cyclers, which allow researchers to test a range of annealing temperatures in a single experiment to empirically determine the ideal conditions for their specific primers and template.
Stage 3: Extension – Synthesizing New DNA
The final stage of the cycle is extension, or elongation. The temperature is raised to the optimal operating temperature of the DNA polymerase, which for the commonly used Taq polymerase is 72°C. At this temperature, the polymerase enzyme binds to the primer-template complex and begins to synthesize a new, complementary strand of DNA. It does this by adding free deoxynucleotide triphosphates (dNTPs) from the reaction mixture, extending from the 3′ end of the primer in a 5′ to 3′ direction. The duration of the extension step is determined by the length of the DNA fragment being amplified, with a general rule of thumb being approximately one minute for every 1,000 base pairs (1 kb) of the target sequence. At the end of this stage, what was one double-stranded DNA molecule at the start of the cycle has become two. These two molecules then serve as templates for the next cycle of denaturation, annealing, and extension, leading to the exponential accumulation of the target DNA.
The Reaction Mixture: Essential Components for PCR
|
|
A successful PCR amplification depends on a precise combination of key reagents, each playing a critical role in the reaction. These components are typically combined in a small reaction tube or a well of a multi-well plate.
- DNA Template: This is the sample containing the target DNA sequence that is to be amplified. The template can be genomic DNA, plasmid DNA, or complementary DNA (cDNA) derived from RNA. The purity and integrity of the template DNA are of utmost importance, as contaminants can inhibit the polymerase enzyme and lead to reaction failure.
- Thermostable DNA Polymerase: This is the enzyme that serves as the master builder, synthesizing the new DNA strands. The discovery of thermostable polymerases was a watershed moment for PCR. The most common is Taq polymerase, isolated from Thermus aquaticus, which is prized for its ability to function at high temperatures. For applications that demand a higher degree of accuracy and fewer copying errors, such as cloning or sequencing, high-fidelity polymerases like Pfu polymerase (from Pyrococcus furiosus) are often used.
- Primers: These are short, chemically synthesized single-stranded DNA fragments, known as oligonucleotides, typically 18 to 30 nucleotides in length. Two distinct primers, a forward primer and a reverse primer, are used in each reaction. They are designed to be complementary to the 3′ ends of the opposing strands of the target DNA, effectively bracketing the specific region to be amplified. The design of these primers is a crucial determinant of the specificity and efficiency of the PCR reaction.
- Deoxynucleotide Triphosphates (dNTPs): These are the fundamental building blocks of DNA. The reaction mixture must contain an adequate supply of all four dNTPs—dATP, dGTP, dCTP, and dTTP—which the DNA polymerase incorporates into the growing DNA strand during the extension phase.
- PCR Buffer and Cofactors (MgCl₂): The reaction is carried out in a buffer solution that maintains an optimal pH and ionic environment for the DNA polymerase to function effectively. A critical component of this buffer system is magnesium chloride (MgCl2). Magnesium ions (Mg2+) act as an essential cofactor for the polymerase enzyme. The concentration of Mg2+ is a critical parameter to optimize, as it influences not only the enzyme’s activity but also primer annealing and the overall specificity of the reaction. Too little magnesium can lead to low yield, while too much can promote non-specific amplification.
Streamlining Workflows: The Role of the PCR Master Mix
To improve efficiency and consistency, many laboratories have moved away from preparing reaction mixtures by adding each component individually. Instead, they utilize commercial PCR master mixes. A master mix is a pre-prepared, concentrated solution that contains the DNA polymerase, dNTPs, MgCl2, and reaction buffer in an optimized formulation. The researcher only needs to add the template DNA, primers, and nuclease-free water to this mix.
The use of master mixes offers significant practical advantages. It dramatically reduces the number of pipetting steps required to set up reactions, which not only saves considerable time but also minimizes the risk of pipetting errors and contamination. This leads to greatly improved consistency and reproducibility of results, both within a single experiment and between different experiments and laboratories. This is particularly valuable in high-throughput applications where hundreds or thousands of reactions are run simultaneously. The widespread adoption of these pre-optimized solutions reflects a broader trend in the life sciences industry toward standardization and quality control. This shift effectively transfers the burden of optimizing the concentrations of core reagents from the individual researcher to the manufacturer, who can produce large, quality-controlled batches. This allows scientists to focus their efforts on the more variable aspects of their experimental design, such as primer specificity and template quality, ultimately leading to more reliable and robust scientific outcomes.
The Workhorse of the Lab: The Thermal Cycler
The automation of the Polymerase Chain Reaction is made possible by a single, indispensable piece of laboratory equipment: the thermal cycler. Also commonly referred to as a PCR machine or DNA amplifier, this instrument is designed to precisely execute the series of temperature cycles required for DNA amplification, following a user-defined program.
Key Components and Their Functions
Modern thermal cyclers are sophisticated instruments built around several core components that work in concert to ensure reliable and efficient PCR.
 |
 |
-
- Thermal Block: This is the heart of the instrument, a precisely machined block, often made of materials with high thermal conductivity like silver or aluminum, with wells to hold the PCR tubes or plates. The temperature of this block is controlled by a Peltier element. This solid-state thermoelectric device enables extremely rapid heating and cooling simply by reversing the direction of an electrical current passing through it, a key feature for minimizing the time spent transitioning between the denaturation, annealing, and extension steps.
 |
- Heated Lid: A standard and essential feature of all modern thermal cyclers, the heated lid presses firmly against the caps of the reaction tubes or the seal of a plate. Its purpose is to maintain a high temperature (typically around 105°C) to prevent the small reaction volumes from evaporating and condensing on the cooler upper surfaces of the tubes during the temperature cycles. This simple innovation eliminated the need for the messy and inconvenient mineral oil overlays that were required in early machines to prevent sample loss.
- User Interface & Software: The instrument’s control system allows users to program, store, and execute specific PCR protocols. Early models had simple keypads and text-based displays, but contemporary thermal cyclers typically feature intuitive, graphical touch-screen interfaces. Advanced software may also offer features like remote monitoring via a network or mobile device and cloud connectivity for protocol sharing and data management.
Choosing the Right Instrument: Key Considerations for Procurement
The specifications of a thermal cycler have a direct and significant impact on both the speed of research and the quality and reproducibility of the results. For lab managers and procurement specialists, selecting the right instrument involves evaluating several key features that go beyond basic functionality.
- Throughput and Flexibility: Thermal cyclers are available in a wide range of formats to suit different throughput needs, from instruments that hold a few individual tubes to those that accommodate 96-well or 384-well plates. For labs with diverse and evolving needs, models with interchangeable thermal blocks offer valuable flexibility, allowing a single instrument to be adapted for different reaction volumes and vessel types.
- Ramp Rate: This specification refers to the speed at which the thermal block can change temperature, typically measured in degrees Celsius per second (°C/sec). A higher ramp rate translates directly to shorter PCR run times, which is a critical factor for increasing throughput in busy labs. It is important to differentiate between the maximum block ramp rate, which may only be achieved for a brief moment, and the average sample ramp rate, which is a more realistic measure of how quickly the actual reaction mixture inside the tube changes temperature.
- Gradient Capabilities: This is a powerful feature for protocol optimization. A gradient thermal cycler can maintain a linear temperature gradient across the thermal block during the annealing step. This allows a researcher to test a range of different annealing temperatures (e.g., from 50°C to 65°C) simultaneously in a single run, making it vastly more efficient to determine the optimal temperature for a new set of primers. Some advanced instruments feature segmented blocks with independent heating and cooling elements, providing even more precise temperature control than a traditional gradient. These features are not mere conveniences; they are scientific tools that enable more robust experimental design and lead to more reliable and reproducible data. For a procurement decision, the higher initial investment in an instrument with advanced temperature control features can yield a significant long-term return through savings in researcher time, expensive reagents, and failed experiments.
The PCR Family: A Guide to Common PCR Techniques
Since its invention, the basic PCR principle has been adapted and modified into a diverse family of specialized techniques. Each variant offers unique capabilities, allowing researchers to ask more sophisticated questions about nucleic acids. Understanding the distinctions between these methods is crucial for selecting the appropriate tool for a given experimental goal.
| Technology | Principle | Starting Material | Key Output | Primary Applications | Advantages | Disadvantages |
| Conventional PCR | End-point amplification of a specific DNA target. | DNA | Qualitative or semi-quantitative presence/absence of an amplicon, visualized on a gel. | Gene cloning, genotyping, sequencing preparation, basic diagnostics. | Simple, robust, widely established. | Not quantitative, requires post-PCR processing, risk of carryover contamination. |
| RT-PCR | Reverse transcription of RNA into cDNA, followed by standard PCR amplification. | RNA | Presence/absence of a specific RNA transcript. | RNA detection, gene expression profiling (qualitative), cDNA library creation. | Enables the study of RNA viruses and gene expression. | Requires an additional enzymatic step; susceptible to RNA degradation. |
| qPCR (Real-Time PCR) | Real-time monitoring of DNA amplification using fluorescent reporters. | DNA or RNA (as RT-qPCR) | Quantitative data (relative or absolute) on the initial amount of target nucleic acid. | Gene expression analysis, viral load quantification, pathogen detection, GMO testing. | Highly sensitive, quantitative, wide dynamic range, closed-tube system reduces contamination. | Requires specialized, more expensive equipment; relative quantification relies on standards or references. |
| dPCR (Digital PCR) | Partitioning of the sample into thousands of individual reactions for absolute end-point quantification. | DNA or RNA (as RT-dPCR) | Absolute count of target molecules (copies/µL) without a standard curve. | Rare mutation detection, copy number variation (CNV), liquid biopsy, viral load monitoring. | Absolute quantification, extreme precision, high tolerance to inhibitors. | Lower throughput, smaller dynamic range than qPCR, higher cost per sample. |
| Multiplex PCR | Simultaneous amplification of multiple different targets in a single reaction tube. | DNA or RNA | Detection of multiple amplicons, often differentiated by size or fluorescent probe color. | Syndromic pathogen panels, forensic STR analysis, genotyping. | Saves time, cost, and precious sample material; high information output per reaction. | Requires complex optimization of primers and reaction conditions to avoid interference. |
| Nested PCR | Two sequential rounds of PCR using two primer sets; the second set is “nested” within the first amplicon. | DNA or RNA | Highly specific and sensitive detection of a target amplicon. | Detection of very low-abundance targets, amplification from degraded DNA, diagnostics. | Greatly increased sensitivity and specificity. | Increased risk of contamination due to sample handling between rounds; more time-consuming. |
Reverse Transcription PCR (RT-PCR): Analyzing RNA
Standard PCR can only amplify DNA. To study RNA molecules, such as messenger RNA (mRNA) for gene expression analysis or the genomes of RNA viruses, an initial step is required. Reverse Transcription PCR (RT-PCR) addresses this by first using an enzyme called reverse transcriptase to synthesize a DNA copy of the RNA template. This newly created DNA molecule is known as complementary DNA (cDNA), which then serves as the template for a standard PCR amplification. RT-PCR can be performed using two different workflows:
- One-Step RT-PCR: In this approach, the reverse transcription and PCR amplification are performed sequentially in a single, closed tube. This method is fast, requires less sample handling, and minimizes the risk of contamination, making it well-suited for high-throughput applications like clinical diagnostics.
- Two-Step RT-PCR: Here, the reverse transcription is performed first, and then an aliquot of the resulting cDNA product is transferred to a separate tube for the PCR reaction. While more time-consuming and with a higher risk of contamination, this method offers greater flexibility. It allows for the optimization of each reaction independently and creates a stable cDNA archive that can be stored and used for multiple future PCR experiments targeting different genes from the same initial RNA sample.
Quantitative Real-Time PCR (qPCR): Measuring DNA as It Amplifies
While conventional PCR provides a qualitative result—the presence or absence of a product at the end of the reaction—quantitative PCR (qPCR) allows for the measurement of the amount of amplified DNA in real-time, as the reaction progresses. This is achieved by incorporating a fluorescent molecule into the reaction whose signal increases in direct proportion to the amount of amplified DNA. A specialized thermal cycler equipped with an optical detection system measures the fluorescence intensity after each cycle. By tracking the cycle at which the fluorescence crosses a certain threshold, one can accurately determine the starting quantity of the target nucleic acid. With its wide dynamic range and high sensitivity, qPCR (often combined with reverse transcription as RT-qPCR) has become the gold standard for applications like quantifying gene expression levels and measuring viral loads.
Digital PCR (dPCR): Absolute Quantification Through Partitioning
Digital PCR (dPCR) represents a third generation of PCR technology that provides absolute quantification of nucleic acids without the need for standard curves or references. The key innovation in dPCR is the partitioning of the sample. The reaction mixture is divided into thousands or even millions of separate, microscopic partitions (such as droplets in an oil emulsion) before amplification begins. This partitioning is done at a dilution where each partition contains, on average, either one or zero target molecules.
A standard end-point PCR is then performed in all partitions simultaneously. After the reaction, each partition is read as either positive (fluorescent) or negative (non-fluorescent). The fraction of positive partitions is then used, with the help of Poisson statistical analysis, to calculate the absolute concentration of the target molecule in the original sample, expressed as copies per microliter. This evolution from the analog signal of qPCR to the binary counting of dPCR represents a fundamental shift from relative to absolute measurement. This leap was driven by the need for greater precision in applications that were pushing the limits of qPCR, such as detecting extremely rare cancer mutations or resolving small differences in gene copy numbers. The development of sophisticated microfluidic and droplet-generation technologies was the engineering breakthrough that made this powerful digital approach a practical reality. Consequently, dPCR is the method of choice for applications demanding the highest sensitivity and precision, including liquid biopsies, copy number variation (CNV) analysis, and the quantification of low-level pathogens.
Multiplex PCR: Analyzing Multiple Targets at Once
Multiplex PCR enhances efficiency by enabling the simultaneous amplification of several different DNA sequences within a single reaction tube. This is accomplished by including multiple unique primer pairs in the reaction mix, with each pair designed to amplify a specific target. This approach provides significant savings in time, cost, and, most importantly, precious sample material. It is widely used in diagnostic settings for syndromic testing, where a panel of potential pathogens causing similar symptoms can be tested for at once, as well as in genotyping and forensic analysis. The main challenge of multiplex PCR is the complex optimization required to ensure all primer pairs work efficiently under a single set of reaction conditions without interfering with each other or forming non-specific products.
Nested PCR: Enhancing Sensitivity and Specificity
Nested PCR is a modification designed to dramatically increase both the sensitivity and specificity of detection, particularly for low-abundance targets. The technique involves two consecutive rounds of PCR. The first round uses an “outer” set of primers to amplify a larger DNA fragment that contains the target sequence. The product from this first reaction is then used as the template for a second round of PCR, which uses a second, “inner” or “nested” set of primers that bind to sites within the first amplicon. Because it is highly improbable that any non-specific product from the first round would also contain the binding sites for the inner primers, this two-step amplification ensures that only the correct target sequence is amplified to a detectable level. This makes nested PCR an invaluable tool for detecting pathogens present in very small numbers or for amplifying DNA from old or degraded samples.
Applications of PCR Across Scientific Disciplines
The transformative impact of PCR is evident in its vast range of applications across nearly every field of biology and medicine. Its core function—the ability to amplify a specific genetic signal from a complex background—has made it an essential tool for discovery, diagnostics, and quality control. At its heart, PCR’s power lies in its capacity to make the invisible both visible and quantifiable. It serves as a universal translator, converting minuscule and specific biological signals—a single viral particle in a patient sample, a trace of DNA at a crime scene, or a subtle change in a gene’s activity—into a robust and analyzable signal that can be easily detected and measured.
In Clinical Diagnostics: Detecting Pathogens and Genetic Disease
In the realm of clinical diagnostics, PCR has become the gold standard for the rapid and sensitive detection of infectious diseases. Its ability to detect the genetic material of a pathogen directly allows for diagnosis in the earliest stages of infection, often before the body has mounted a measurable immune response. This has been critical in managing diseases like HIV, hepatitis, HPV, and, most notably, in the global response to the COVID-19 pandemic. Beyond infectious diseases, PCR is used to identify specific genetic mutations associated with hereditary disorders and to diagnose and monitor cancer. It can detect cancer-specific DNA sequences or residual cancer cells with a sensitivity that far surpasses traditional methods. Furthermore, the advent of multiplex PCR has enabled syndromic testing, where a single patient sample can be screened for a comprehensive panel of pathogens known to cause similar clinical symptoms, such as respiratory or gastrointestinal infections. This allows clinicians to make faster, more targeted treatment decisions.
In Forensic Science: DNA Fingerprinting and Identification
PCR is the cornerstone of modern forensic science. Its extraordinary sensitivity makes it possible to generate a DNA profile from minute quantities of biological evidence—such as a single hair, a drop of blood, or trace skin cells left on an object—that would be insufficient for older methods of analysis. Forensic analysts use PCR to amplify specific, highly variable regions of the human genome known as Short Tandem Repeats (STRs). The combination of STR markers amplified from a sample creates a unique genetic “fingerprint” that can be compared to suspects or entered into national databases like the FBI’s Combined DNA Index System (CODIS). The landmark case of Colin Pitchfork in the 1980s, who became the first person convicted of murder based on DNA evidence, powerfully demonstrated the potential of these techniques and forever changed the field of criminal investigation.
In Research and Biotechnology: Cloning, Sequencing, and Gene Expression
Within the research laboratory, PCR is a daily workhorse and a fundamental first step in countless molecular biology workflows. It is routinely used to amplify a specific gene of interest from a complex genome. This amplified DNA can then be inserted, or cloned, into a plasmid vector for further study, such as producing large quantities of a specific protein. To facilitate this cloning process, short sequences corresponding to restriction enzyme recognition sites are often added to the 5′ ends of the PCR primers. The amplicons can also be sent for DNA sequencing to determine their exact nucleotide code, a critical step in gene discovery and characterization. In the field of functional genomics, RT-qPCR is the most widely used method for gene expression analysis, allowing scientists to precisely measure how the activity levels of thousands of genes change in response to different conditions, treatments, or diseases.
In Food Safety: Ensuring the Integrity of the Supply Chain
The speed and specificity of PCR have also made it an invaluable tool for the food industry. It is used for the rapid detection of foodborne pathogens like Salmonella, Listeria, and E. coli in raw ingredients and finished products. Compared to traditional microbiological culture methods, which can take days, PCR-based tests can provide results in a matter of hours, enabling faster product release and quicker intervention in the event of contamination. Beyond pathogen detection, PCR is applied for a wide range of quality control and authenticity testing. It can be used to verify product claims, such as detecting the presence of genetically modified organisms (GMOs) in a product labeled as GMO-free, screening for common food allergens, or identifying the species of origin in processed meat products to prevent fraudulent labeling.
Best Practices for Reliable and Reproducible PCR
The extraordinary sensitivity of PCR is both its greatest strength and its greatest challenge. To generate trustworthy and reproducible results, laboratory professionals must adhere to a strict set of best practices covering everything from experimental design to laboratory setup and execution. Effective PCR is a discipline, not just a procedure. This discipline is rooted in the understanding that the biggest threat to a PCR experiment is often the result of a previous successful experiment. The power of PCR to create billions of copies of a target sequence means that the final product, the amplicon, is an extremely potent contaminant for any future reactions targeting the same sequence. This paradox—that the technique’s success is also its greatest liability—necessitates the meticulous, almost ritualistic, practices for contamination control that define high-quality molecular work.
Primer Design: Guidelines for Specificity and Efficiency
The success of a PCR reaction begins with well-designed primers. The following guidelines are critical for ensuring specific and efficient amplification:
- Length: Primers should typically be between 18 and 30 nucleotides long. This length is sufficient to ensure they bind to a unique site in a complex genome while still allowing for efficient annealing.
- Melting Temperature (Tm): The Tm is the temperature at which half of the primer-template duplexes dissociate. For optimal performance, the Tm of both the forward and reverse primers should be between 50°C and 65°C and should not differ from each other by more than 5°C. This ensures that both primers bind with similar efficiency at the chosen annealing temperature.
- GC Content: The percentage of guanine (G) and cytosine (C) bases in a primer should be between 40% and 60%. Because G-C pairs are held together by three hydrogen bonds (compared to two for A-T pairs), a balanced GC content contributes to stable primer binding. Including one or two G or C bases at the 3′ end of the primer, known as a “GC clamp,” can further promote stable binding and improve polymerase efficiency.
- Avoiding Secondary Structures: Primers must be checked to ensure they do not have sequences that are complementary to themselves or to the other primer in the pair. Such complementarity can lead to the formation of internal “hairpin” loops or “primer-dimers,” which can compete with and inhibit the amplification of the desired target. Publicly available online tools, such as Primer-BLAST, should be used to verify both the specificity of the primers and their potential for forming these undesirable structures.
Lab Setup: Aseptic Technique and Preventing Contamination
Because PCR can amplify a single molecule of contaminating DNA, preventing contamination is paramount. The golden rule is to maintain a unidirectional workflow with physically separated areas and dedicated equipment.
- Physical Separation: A PCR workflow should be divided into at least three distinct areas:
- A “clean” pre-PCR area for reagent preparation and master mix assembly.
- A separate area for adding the DNA template to the reaction mixes.
- A “dirty” post-PCR area for thermal cycling and analysis of the amplified products (e.g., gel electrophoresis).Critically, equipment, reagents, and lab coats should never move from a post-PCR area back into a pre-PCR area to prevent carryover contamination of amplicons.
- Dedicated Equipment and Consumables: Each designated area should have its own set of dedicated equipment, including pipettes, tube racks, and vortexers. The use of aerosol-resistant filter pipette tips is essential to prevent the transfer of contaminants via aerosols during pipetting.
- Decontamination: Work surfaces and equipment should be cleaned regularly with a solution that degrades DNA. A freshly prepared 10% bleach solution is effective, as are commercial DNA-degrading solutions. It is important to note that 70% ethanol is not sufficient to remove or destroy nucleic acid contaminants. Enclosed workstations, such as PCR hoods equipped with UV lamps, can also be used to decontaminate surfaces and reagents before use.
- Controls: Every PCR run must include a No Template Control (NTC). This is a reaction that contains all the necessary components except for the DNA template (nuclease-free water is added instead). If the NTC shows an amplification product, it is a clear indication of reagent or environmental contamination, and the results of the entire run should be considered invalid.
Common PCR Problems and Solutions
Even with careful planning, PCR experiments can sometimes fail or produce ambiguous results. The following table outlines common problems and provides recommended troubleshooting steps.
| Problem | Possible Causes | Recommended Solutions |
| No Amplification or Faint Band | – Incorrect annealing temperature (too high). – Poor template quality or presence of inhibitors. – Insufficient number of cycles. – Error in reaction setup (e.g., missing component). – Degraded reagents (polymerase, dNTPs). |
– Optimize annealing temperature using a gradient PCR. – Re-purify template DNA; check A260/280 ratio. – Increase the number of cycles in increments of 5. – Repeat setup carefully; use a checklist. – Use fresh reagents and aliquot to avoid freeze-thaw cycles. |
| Non-Specific Bands (Multiple Bands) | – Annealing temperature is too low. – Primer design is not specific enough. – Too much template or primer concentration. – Too many cycles. – High Mg2+ concentration. |
– Increase annealing temperature in 2°C increments. – Redesign primers; use Primer-BLAST to check specificity. – Titrate template and primer concentrations downwards. – Reduce the number of cycles. – Titrate Mg2+ concentration downwards. |
| Primer-Dimers | – High primer concentration. – Primer design includes 3′ complementarity. – Low annealing temperature. – Reaction setup at room temperature (non-hot-start polymerase). |
– Reduce primer concentration. – Redesign primers to avoid 3′ complementarity. – Increase annealing temperature. – Use a hot-start DNA polymerase and set up reactions on ice. |
| Smear on Gel | – Too much template DNA. – Template DNA is degraded. – Too many cycles. – Annealing temperature is too low. – Extension time is too long. |
– Reduce the amount of template DNA. – Check template integrity on a gel; prepare fresh template. – Reduce the number of cycles. – Increase the annealing temperature. – Reduce the extension time. |
The Importance of High-Quality Reagents and Consumables
Finally, the foundation of any reliable and reproducible PCR experiment is the quality of the materials used. The purity and performance of the DNA polymerase, dNTPs, primers, and even the plastic consumables (tubes and plates) directly influence the outcome. Low-grade or contaminated reagents can introduce variability, inhibit the reaction, and lead to inconsistent or entirely false results, wasting valuable time, samples, and resources. Sourcing certified, high-purity reagents and consumables from trusted manufacturers is a critical investment in the integrity and validity of the scientific data generated.
The Future of PCR: Trends and Innovations
Since its inception, PCR technology has been in a state of continuous evolution. Looking forward, several key trends are poised to further expand its capabilities and applications, pushing diagnostics and research into new frontiers. The future trajectory of PCR is being defined by two powerful, converging forces: decentralization of the technology and integration of the data it generates. These trends are transforming PCR from a tool used by specialists in centralized laboratories into an accessible, networked instrument with the potential to impact population-level health.
Miniaturization and Point-of-Care (POC) Diagnostics
A dominant trend is the miniaturization of PCR technology into “lab-on-a-chip” systems. These microfluidic devices integrate sample preparation, amplification, and detection into a single, portable, and often automated instrument. This technological shift is driving the decentralization of molecular diagnostics, moving testing from large, central labs to the point of care—be it a doctor’s office, a remote clinic, or even in the field for environmental monitoring or on-site forensic analysis. The global COVID-19 pandemic dramatically accelerated the development, regulatory approval, and adoption of such POC systems, demonstrating their immense value in rapid response scenarios and setting a clear precedent for the future of infectious disease diagnostics.
The Rise of Digital PCR in Clinical Applications
As the third generation of PCR, digital PCR is rapidly moving from a specialized research tool to a clinical workhorse. Its unparalleled precision, sensitivity, and resistance to common PCR inhibitors make it uniquely suited for challenging clinical applications where qPCR may fall short. dPCR is becoming the method of choice for non-invasive liquid biopsies to detect and monitor circulating tumor DNA in cancer patients, for tracking minimal residual disease after treatment, and for providing highly accurate viral load measurements for managing chronic infections like HIV and hepatitis.
Integration with Automation and Advanced Data Analysis
To meet the demands of large-scale research and clinical testing, high-throughput PCR systems are increasingly being integrated with robotic liquid-handling platforms. This full automation of the workflow minimizes human error, increases throughput, and ensures a higher degree of standardization and reproducibility. In parallel, the vast amounts of data generated by these systems, particularly from qPCR and dPCR, are being managed by sophisticated software and cloud-based platforms. This allows for advanced data analysis, sharing, and trend monitoring. This data integration holds the potential to connect disparate diagnostic systems into a global “internet of things.” For example, networked dPCR systems monitoring wastewater could provide public health officials with a real-time map of emerging viral outbreaks, enabling faster, more targeted interventions and transforming PCR into a powerful tool for global health surveillance.
Discover HINOTEK full lineup of advanced PCR.
Works cited
- POLYMERASE CHAIN REACTION: METHODS, PRINCIPLES AND APPLICATION – Semantic Scholar, https://pdfs.semanticscholar.org/7925/d6d088c6eacc7e19dbce0e69fd56a7cd4188.pdf
- PCR-based Diagnostics for Infectious Diseases, Genetic Disorders, and Cancer – MyBioSource Learning Center, https://www.mybiosource.com/learn/pcr-based-diagnostics-for-infectious-diseases-genetic-disorders-and-cancer/
- Comprehensive Guide to Real-Time PCR for Food Safety Testing – Hygiena, https://www.hygiena.com/learning-center/technology-guide/comprehensive-guide-to-real-time-pcr-for-food-safety-testing
- Thermal cycler – Wikipedia, https://en.wikipedia.org/wiki/Thermal_cycler
- PCR Thermal Cyclers Education | Thermo Fisher Scientific – US, https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-thermal-cyclers.html
- Thermal Cyclers – The Lab World Group, https://www.thelabworldgroup.com/blog/all-about-thermal-cyclers/
- Introduction to PCR Reagents and Purification – Bio-Rad, https://www.bio-rad.com/en-us/applications-technologies/introduction-pcr-reagents-purification?ID=LUSO0V4VY
- PCR Machine: Principle, Parts, Steps, Types, Uses, Examples – Microbe Notes, https://microbenotes.com/pcr-machine-principle-parts-steps-types-uses-examples/
This guide is maintained by HINOTEK’s core technical team, comprised of senior engineers and application scientists with over two decades of hands-on experience in fields such as microscopy, centrifugation, and spectrophotometry. We are committed to ensuring that every piece of information in this guide—from instrument principles and technical specifications to laboratory procurement advice—maintains the highest level of accuracy and timeliness.
This content is regularly reviewed and updated to reflect the latest industry standards and technological advancements. We value feedback from the global scientific community. Should you have any questions or suggestions, or wish to discuss any technical details, please do not hesitate to contact our expert team at [email protected].